Nerve Blocks App
Nerve Blocks App Pain Medicine Assistant App
Pain Medicine Assistant App POCUS App
POCUS App IV Access App
IV Access App MSK Knee App
MSK Knee App VetRA App
VetRA App Nerve Block Manual
Nerve Block Manual Regional Anesthesia Updates
Regional Anesthesia Updates Anesthesiology Manual
Anesthesiology Manual Anesthesiology Review
Anesthesiology Review Anesthesia Updates 2025
Anesthesia Updates 2025 Anesthesia Updates 2026
Anesthesia Updates 2026 Pediatric Anesthesia Updates
Pediatric Anesthesia Updates Airway Management Updates
Airway Management Updates US Interventional Pain Manual
US Interventional Pain Manual Pain Medicine Updates
Pain Medicine Updates Mastering Difficult IV Access
Mastering Difficult IV Access PACU Nursing Manual
PACU Nursing Manual RA Veterinary Manual
RA Veterinary Manual About
About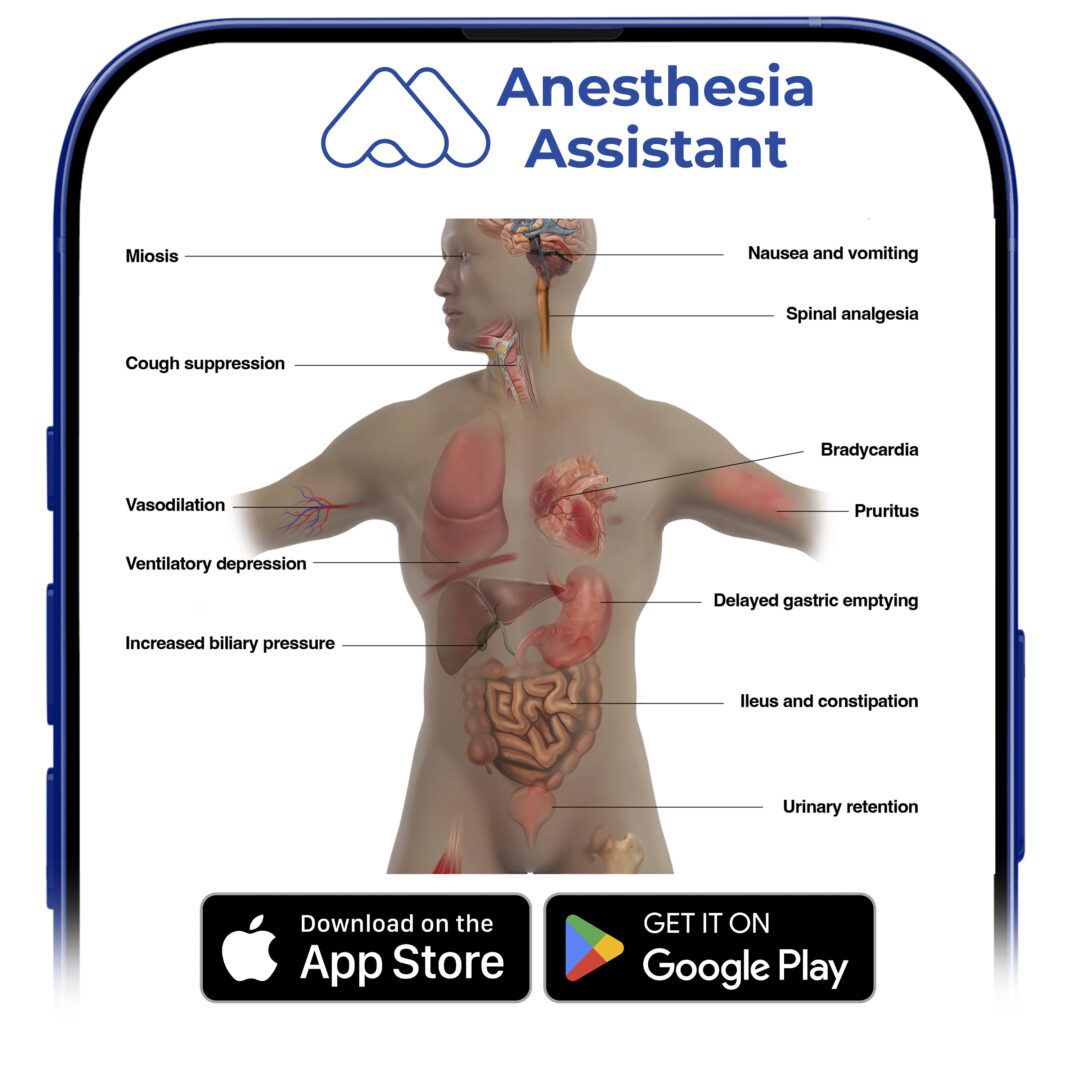
Fentanyl, remifentanil, or morphine?
Compare and dose opioids instantly with the Anesthesia Assistant App.
Opioids play an indispensable role in the practice of anesthesiology, critical care, and pain management. A sound understanding of opioid pharmacology, including both basic science and clinical aspects, is critical for the safe and effective use of these important drugs. This chapter will focus almost exclusively on intravenous opioid receptor agonists used perioperatively.
1. BASIC PHARMACOLOGY
Structure-Activity
The opioids of clinical interest in anesthesiology share many structural features. Morphine is a benzylisoquinoline alkaloid (Fig.1). Many commonly used semisynthetic opioids are created by simple modification of the morphine molecule. Codeine, for example, is the 3-methyl derivative of morphine. Similarly, hydromorphone, hydrocodone, and oxycodone are also synthesized by relatively simple modifications of morphine. More complex alterations of the morphine molecular skeleton result in mixed agonist-antagonists such as nalbuphine and even complete antagonists such as naloxone.
The fentanyl series of opioids are chemically related to meperidine. Meperidine is the first completely synthetic opioid and can be regarded as the prototype clinical phenylpiperidine (see Fig 1). Fentanyl is a simple modification of the basic phenylpiperidine structure. Other commonly used fentanyl congeners such as alfentanil and sufentanil are somewhat more complex versions of the same phenylpiperidine skeleton.
Opioids share many physicochemical features in common, although some individual drugs have unique features. In general, opioids are highly soluble weak bases that are highly protein bound and largely ionized at physiologic pH. Opioid physicochemical properties influence their clinical behavior. For example, relatively unbound, unionized molecules such as alfentanil and remifentanil have a shorter latency to peak effect after bolus injection.
Mechanism
Opioids produce their main pharmacologic effects by interacting with opioid receptors, which are typical of the G protein–coupled family of receptors widely found in biology (e.g., β-adrenergic, dopaminergic, among others). Expression of cloned opioid receptors in cultured cells has facilitated analysis of the intracellular signal transduction mechanisms activated by the opioid receptors.(1) Binding of opioid agonists with the receptors leads to activation of the G protein, producing effects that are primarily inhibitory (Fig. 2); these effects ultimately culminate in hyperpolarization of the cell and reduction of neuronal excitability.
Three classical opioid receptors have been identified using molecular biology techniques: μ, κ, and δ. More recently, a fourth opioid receptor, ORL1 (also known as NOP), has also been identified, although its function is quite different from that of the classical opioid receptors. Each of these opioid receptors has a commonly employed experimental bioassay, associated endogenous ligand(s), a set of agonists and antagonists, and a spectrum of physiologic effects when the receptor is agonized. Although the existence of opioid receptor subtypes (e.g., μ1 μ2) has been proposed, it is not clear from molecular biology techniques that distinct genes exist for them. Posttranslational modification of opioid receptors certainly occurs and may be responsible for conflicting data regarding opioid receptor subtypes.(2)
Opioids exert their therapeutic effects at multiple sites. They inhibit the release of substance P from primary sensory neurons in the dorsal horn of the spinal cord, mitigating the transfer of painful sensations to the brain. Opioid actions in the brainstem modulate nociceptive transmission in the dorsal horn of the spinal cord through descending inhibitory pathways. Opioids are thought to change the affective response to pain through actions in the forebrain; decerebration prevents opioid analgesic efficacy in rats.(3) Furthermore, morphine induces signal changes in “reward structures” in the human brain.(4)
Studies in genetically altered mice have yielded important information about opioid receptor function. In μ opioid receptor knockout mice, morphine-induced analgesia, reward effect, and withdrawal effect are absent.(5,6) Importantly, μ receptor knockout mice also fail to exhibit respiratory depression in response to morphine.(7)
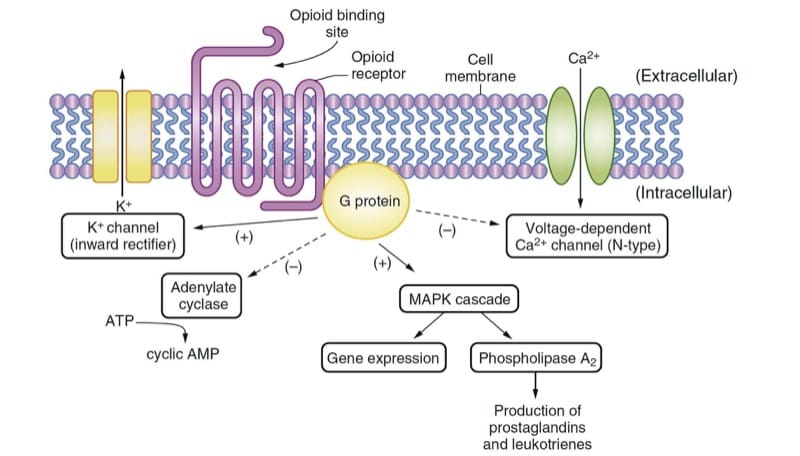
- Fig.2 Opioid mechanisms of action. The endogenous ligand or drug binds to the opioid receptor and activates the G protein, resulting in multiple effects that are primarily inhibitory. The activities of adenylate cyclase and the voltage-dependent Ca2+ channels are depressed. The inwardly rectifying K+ channels and mitogen activated protein kinase (MAPK) cascade are activated. AMP, Adenosine monophosphate; ATP, adenosine triphosphate.
Metabolism
The intravenously administered opioids in routine perioperative clinical use are transformed and excreted by many metabolic pathways. In general, opioids are metabolized by the hepatic microsomal system, although hepatic conjugation and subsequent excretion by the kidney are important for some opioids. For certain opioids, the specific metabolic pathway involved has important clinical implications in terms of active metabolites (e.g., morphine, meperidine) or an ultra short duration of action (e.g., remifentanil). For other opioids, genetic variation in the metabolic pathway can drastically alter the clinical effects (e.g., codeine). These nuances are addressed in a subsequent section focused on individual drugs.
2. CLINICAL PHARMACOLOGY
Pharmacokinetics
Pharmacokinetic differences are the primary basis for the rational selection and administration of opioids in perioperative anesthesia practice. Key pharmacokinetic behaviors are (1) the latency to peak effect-site concentration after bolus injection (i.e., bolus front-end kinetics), (2) the time to clinically relevant decay of concentration after bolus injection (i.e., bolus back-end kinetics), (3) the time to steady-state concentration after starting a continuous infusion (i.e., infusion front-end kinetics), and (4) the time to clinically relevant decay in concentration after stopping a continuous infusion (i.e., infusion back-end kinetics).
Applying opioid pharmacokinetic concepts to clinical anesthesiology requires recognition of several fundamental principles. First, a table of pharmacokinetic variables has limited clinical value. Understanding pharmacokinetic behavior is best achieved through computer simulation. Second, opioids administered by bolus injection or continuous infusion must be considered separately. (8) Third, pharmacokinetic information must be integrated with knowledge about the concentration-effect relationship and drug interactions (i.e., pharmacodynamics) in order to be clinically useful.
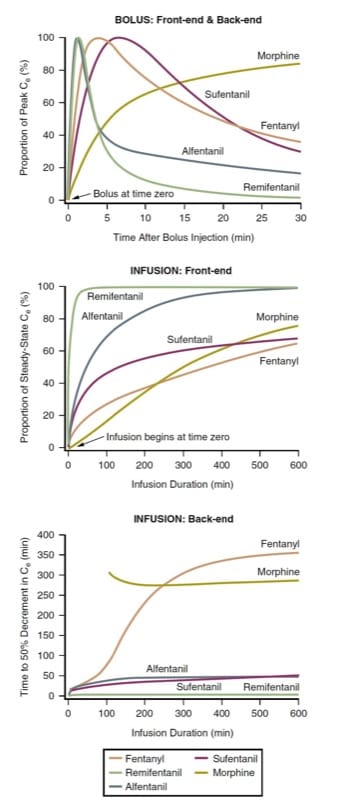
The latency to peak effect and the offset of effect after bolus injection (i.e., bolus front-end kinetics and bolus back-end kinetics) of various intravenous opioids can be defined by predicting the time course of effect-site concentrations after a bolus is administered. Because the opioids differ in terms of potency (and thus the required dosages), for comparison purposes, the effect-site concentrations must be normalized to the percent of peak concentration for each drug. Considering morphine, fentanyl, sufentanil, alfentanil, and remifentanil as among the opioids most commonly used intraoperatively, pharmacokinetic simulation illustrates how opioids differ in terms of latency to peak effect after a bolus is adminis-tered (Fig.3, top panel). (9-12)
The simulation of a bolus injection (see Fig.3, top panel) has clinical implications. For example, when a rapid onset of opioid effect is desirable, morphine may not be a good choice. Similarly, when the clinical goal is a brief duration of opioid effect followed by rapid dissipation, remifentanil or alfentanil might be preferred. Note how remifentanil’s concentration has declined very substantially before fentanyl’s peak concentration has even been achieved. The simulation illustrates why the front-end kinetics of fentanyl make it a drug well suited for patient-controlled analgesia (PCA) (also see Chapters Postanesthesia Recovery and Perioperative Pain Manageme). In contrast to morphine, the peak effect of a fentanyl bolus is manifest before a typical PCA lock-out period has elapsed, thus mitigating a “dose stacking” problem (also see Chapter Perioperative Pain Management).
The latency to peak effect is governed by the speed with which the plasma and effect site come to equilibrium (i.e., the ke0 parameter). Drugs with a more rapid equilibration have a higher “diffusible” fraction (i.e., the proportion of drug that is unionized and unbound) and high lipid solubility. However, a very large dose of even a slow onset opioid can produce an apparent rapid onset (because a supratherapeutic drug level in the effect site is reached even though the peak concentration comes later).
This simulation of simple, constant rate infusions has obvious clinical implications. First, the time required to reach a substantial fraction of the ultimate steady-state concentration is very long in the context of intraoperative use. To reach a near steady-state more quickly requires that a bolus be administered before the infusion is commenced (or increased). Remifentanil perhaps represents a partial exception to this general rule. Also, opioid concentrations will increase for many hours after an infusion is commenced; in other words, concentrations are typically increasing even though the infusion rate may have been the same for hours! That remifentanil achieves a near steady-state relatively quickly is certainly part of why it has emerged as a popular drug for total intravenous anesthesia (TIVA).
The time to steady-state after beginning a continuous infusion is also best examined by pharmacokinetic simulation. Using the same prototypes as with bolus administration, pharmacokinetic simulation (Fig. 3, middle panel) shows the time required to achieve steady-state effect-site concentrations (i.e., infusion front-end kinetics).
The time to offset of effect after stopping a steady-state infusion is best expressed by the context-sensitive half-time (CSHT) simulation.(13) Defined as the time required to achieve a 50% decrease in concentration after stopping a continuous, steady-state infusion, the CSHT is a means of normalizing the pharmacokinetic behavior of drugs so that rational comparisons can be made regarding the predicted offset of drug effect. The CSHT is thus focused on “infusion back-end” kinetics.
The bottom panel of (Fig.3) is a CSHT simulation for commonly used opioids. For most drugs, the CSHT changes with time. Thus, for brief infusions, the predicted back-end kinetics for the various drugs do not differ much (remifentanil is a notable exception to this general rule). As the infusion time lengthens, the CSHTs begin to differentiate, providing a rational basis for drug selection. Second, depending on the desired duration of opioid effect, either shorter-acting or longer-acting drugs can be chosen. Finally, the shapes of these curves differ depend-ing on the degree of concentration decline required. In other words, the curves representing the time required to achieve a 20% or an 80% decrease in concentration (e.g., the 20% or 80% decrement time simulations) are quite different.(8) Thus, depending on the anesthesia technique applied, the CSHT simulations are not necessarily the clinically relevant simulations (i.e., a 50% decrease may not be the clinical goal). Also, CSHT simulation for morphine does not account for active metabolites (see later discussion of individual drugs under “Unique Features of Individual Opioids”).

- Fig.3 Opioid pharmacokinetics. Simulations illustrating front-end and back-end pharmacokinetic behavior after administration by bolus injection or continuous infusions of morphine, fentanyl, alfentanil, sufentanil, and remifentanil using pharmacokinetic parameters from the literature (see text for details).(9-12,45)
Pharmacodynamics
In most respects, the μ-agonist opioids can be considered pharmacodynamic equals with important pharmacokinetic differences; that is, both the therapeutic and adverse effects are essentially the same. Their efficacy as analgesics and their propensity to produce ventilatory depression are indistinguishable from each other. Pharmacodynamic differences do exist with nonopioid receptor mechanisms such as histamine release.
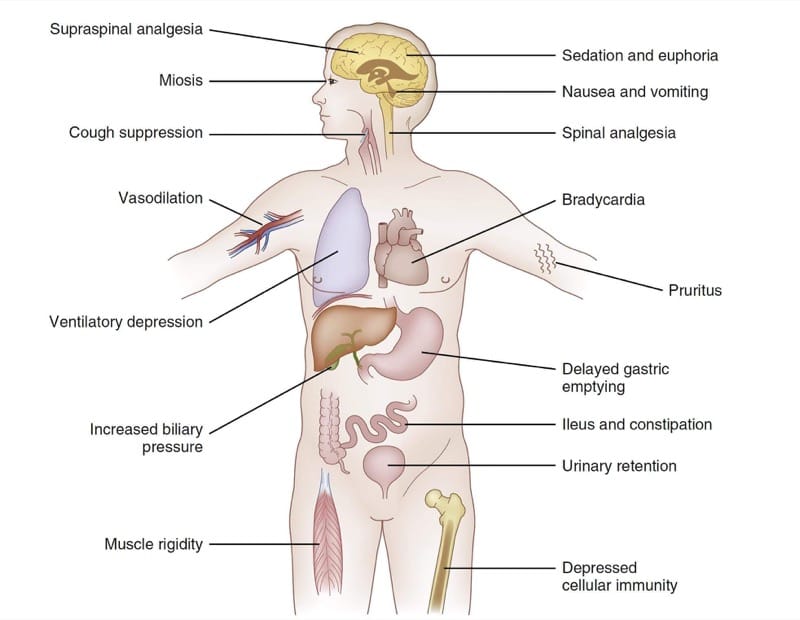
Because the nervous system profoundly influences the function of the entire body, opioid μ-agonist pharmacodynamic effects are observed in many organ systems. Fig.4 summarizes the major pharmacodynamic effects of the fentanyl congeners. Depending on the clinical circumstances and clinical goals of treatment, some of these widespread effects can be viewed as therapeutic or adverse. For example, in some clinical settings the sedation produced by μ-agonists might be viewed as a goal of therapy. In others, drowsiness would clearly be thought of as an adverse effect.

- Fig. 4 Opioid pharmacodynamics. A summary chart of selected effects of the fentanyl congeners (see text for details).
Therapeutic Effects
The relief of pain is the primary therapeutic effect of opioid analgesics. Acting at spinal and brain μ-receptors, opioids provide analgesia both by attenuating the nociceptive traffic from the periphery and also by altering the affective response to painful stimulation centrally.
μ-Agonists are most effective in treating “second pain” sensations carried by slowly conducting, unmyelinated C fibers; they are less effective in treating “first pain” sensations (carried by small, myelinated A-delta fibers) and neuropathic pain. A unique aspect of opioid-induced analgesia (in contrast to drugs like local anesthetics) is that other sensory modalities are not affected (e.g., touch, temperature, among others).
Perioperatively (certainly intraoperatively), the drowsiness produced by μ-agonists is also one of the targeted effects. The brain is the anatomic substrate for the sedative action of μ-agonists. With increasing doses, μ-agonists eventually produce drowsiness and sleep (the relief of pain no doubt contributes to the promotion of sleep in uncomfortable patients both pre- and postoperatively). With sufficient doses, the μ-agonists produce pronounced delta wave activity on the electroencephalogram, which resembles the pattern observed during natural sleep.
μ-Agonists can of course produce significant relief of pain by doses that do not produce sleep. This is the clinical basis for their use in the treatment of pain in ambulatory patients. Yet, the administration of additional doses eventually produces drowsiness (and, as a consequence, the inability to request additional doses) and is the essential scientific foundation for the safety of PCA devices (also see Chapter perioperative pain management). However, even large doses of opioids do not reliably produce unresponsiveness and amnesia and thus opioids cannot be viewed as complete anesthetics when used alone.
Opioids also suppress the cough reflex via the cough centers in the medulla. Attenuation of the cough reflex presumably makes coughing and “bucking” against the indwelling endotracheal tube less likely.
Adverse Effects

Depression of ventilation is the primary adverse effect associated with μ-agonist drugs. When the airway is secured and ventilation is controlled intraoperatively, opioid-induced depression of ventilation is of little consequence. However, opioid-induced respiratory depression in the postoperative period can lead to brain injury and death.
μ-Agonists alter the ventilatory response to arterial carbon dioxide concentrations at the ventilatory control center in the medulla. The depression of ventilation is mediated by the μ-receptor; μ-receptor knock-out mice do not exhibit respiratory depression from morphine.(14)
In unmedicated humans, increases in arterial carbon dioxide partial pressure markedly increase minute volume (Fig.5). Under the influence of opioid analgesics, the curve is flattened and shifted to the right for a given carbon dioxide partial pressure and reflecting that the minute volume is smaller.(15) More importantly, the “hockey stick” shape of the normal curve is lost; that is, there may be a partial pressure of carbon dioxide below which the patient will not breathe (i.e., the “apneic threshold”) in the presence of opioids.

- Fig.5 Opioid-induced ventilatory depression study methodology. The method characterizes the relationship between Paco2 and minute volume. The curve labeled “Normal” represents the expected response of minute volume to rising Paco2 levels in an awake human. Note the dramatic increase in minute volume as CO2 tension rises. The curve labeled “Opioid” represents the blunted response of minute volume to rising CO2 levels following administration of an opioid. Note that the slope of the curve decreases and the curve no longer has a “hockey stick” shape; this means that at physiologic Paco2 levels, the patient receiving sufficient opioid may be apneic or severely hypoventilatory. (Adapted from Gross JB. When you breathe IN you inspire, when you DON’T breathe, you . . . expire: new insights regarding opioid-induced ventila-tory depression. Anesthesiology. 2003;99:767-770, used with permission.)
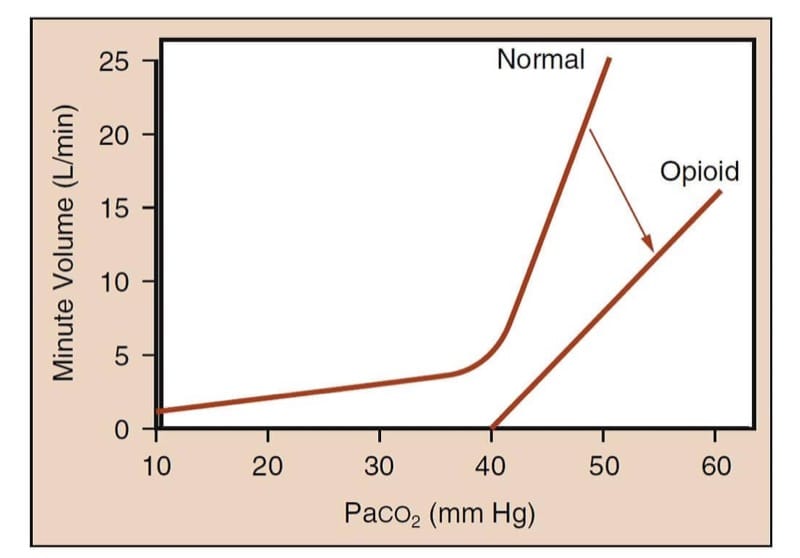
The clinical signs of depressed ventilation are quite subtle with moderate opioid doses. Postoperative patients receiving opioid analgesic therapy can be awake and alert and yet have a significantly decreased minute volume. Respiratory rate (often associated with a slightly increased tidal volume) also decreases. As the opioid concentration is increased, the respiratory rate and tidal volume progressively decrease, eventually culminating in an irregular ventilatory rhythm and then complete apnea.
Many factors can increase the risk of opioid-induced ventilatory depression. Clear risk factors include large opioid dose, advanced age, concomitant use of other central nervous system (CNS) depressants, and renal insufficiency (for morphine). Natural sleep also increases the ventilatory depressant effect of opioids.(16)
Opioids can alter cardiovascular physiology by a variety of different mechanisms. Compared to many other anesthetic drugs (e.g., propofol, volatile anesthetics), however, the cardiovascular effects of opioids, particularly the fentanyl congeners, are relatively minimal (morphine and meperidine are exceptions—see the following section on individual drugs).
The fentanyl congeners cause bradycardia by directly increasing vagal nerve tone in the brainstem, which experimentally can be blocked by microinjection of naloxone into the vagal nerve nucleus or by peripheral vagotomy.(17,18)
Opioids also produce vasodilation by depressing vasomotor centers in the brainstem and to a lesser extent by a direct effect on vessels. This action decreases both preload and afterload. Decreases in arterial blood pressure are more pronounced in patients with increased sympathetic tone such as patients with congestive heart failure or hypertension. Clinical doses of opioids do not appreciably alter myocardial contractility.
Opioids can induce muscle rigidity, usually from the rapid administration of large bolus doses of the fentanyl congeners. This rigidity can even make ventilation via a bag and mask during induction of anesthesia nearly impossible because of vocal cord rigidity and closure.(19) The appearance of rigidity tends to coincide with the onset of unresponsiveness.(20) Although the mechanism of opioid-induced muscle rigidity is unknown, it is not a direct action on muscle because it can be eliminated by the administration of neuromuscular blocking drugs.
Pupillary constriction induced by μ-agonists can be a useful diagnostic sign indicating some ongoing opioid effect. Opioids stimulate the Edinger-Westphal nucleus of the oculomotor nerve to produce miosis. Even small doses of opioid elicit this response and very little tolerance to the effect develops. Thus, miosis is a useful, albeit nonspecific indicator of opioid exposure even in opioid-tolerant patients. Opioid-induced pupillary constriction is naloxone reversible.
Opioids have important effects on gastrointestinal physiology. Opioid receptors are located throughout the enteric plexus of the bowel. Stimulation of these receptors by opioids causes tonic contraction of gastrointestinal smooth muscle, thereby decreasing coordinated, peristaltic contractions. Clinically, this contraction results in delayed gastric emptying and presumably larger gastric volumes in patients receiving opioid therapy preoperatively. Postoperatively, patients can develop opioid-induced ileus that can potentially delay the resumption of proper nutrition and discharge from the hospital. An extension of this acute problem is the chronic constipation associated with long-term opioid therapy.
Similar effects are observed in the biliary system, which also has an abundance of μ-receptors. μ-Agonists can produce contraction of the gallbladder smooth muscle and spasm of the sphincter of Oddi, potentially causing a falsely positive cholangiogram during gallbladder and bile duct surgery. These effects are completely naloxone reversible and can be partially reversed by glucagon treatment.
Although the urologic effects are minimal, opioids can sometimes cause urinary retention by decreasing bladder detrusor tone and by increasing the tone of the urinary sphincter. These effects are in part centrally mediated, although peripheral effects are also likely given the widespread presence of opioid receptors in the genitourinary tract.(21,22) Although the urinary retention associated with opioid therapy is not typically pronounced, it can be troublesome in males, particularly when the opioid is administered intrathecally or epidurally.
Opioids depress cellular immunity. Morphine and the endogenous opioid β-endorphin, for example, inhibit the transcription of interleukin 2 in activated T cells, among other immunologic effects.(23) Individual opioids (and perhaps classes of opioids) may differ in terms of the exact nature and extent of their immunomodulatory effects. Although opioid-induced impairment of cellular immunity is not well understood, impaired wound healing, perioperative infections, and cancer recurrence are possible adverse outcomes.
Drug Interactions
Drug interactions can be based on two mechanisms: pharmacokinetic (i.e., when one drug influences the concentration of the other) or pharmacodynamic (i.e., when one drug influences the effect of the other). In anesthesia practice, although unintended pharmacokinetic interactions sometimes occur, pharmacodynamic interactions occur with virtually every anesthetic and are often produced by design.
The most common pharmacokinetic interaction in opioid clinical pharmacology is observed when intravenous opioids are combined with propofol. Perhaps because of the hemodynamic changes induced by propofol and their impact on pharmacokinetic processes, opioid concentrations may be larger when given in combination with a continuous propofol infusion.(24)
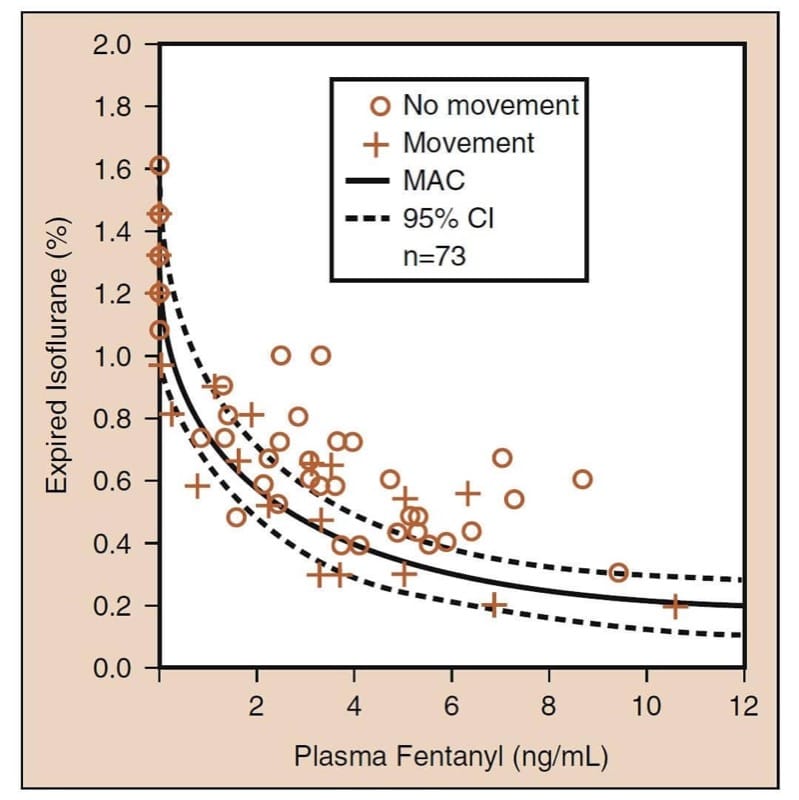
The most important pharmacodynamic drug interaction involving opioids is the synergistic interaction that occurs when opioids are combined with sedatives. (25) When combined with volatile anesthetics, opioids reduce the minimum alveolar concentration (MAC) of a volatile anesthetic (Fig.6). Careful examination of “opioid-MAC reduction” data reveals several clinically critical concepts (see Fig.6). First, opioids synergistically reduce MAC. Second, the MAC reduction is substantial (as much as 75% or more). Third, most of the MAC reduction occurs at moderate opioid levels (i.e., even modest opioid doses substantially reduce MAC). Fourth, reduction of MAC is not complete (i.e., opioids are not complete anesthetics). The addition of the opioid cannot completely eliminate the need for the other anesthetic. And fifth, there are an infinite number of hypnotic-opioid combinations that will achieve MAC (this implies that clinicians must choose the optimal combination based on the goals of the anesthetic and operation). All of these concepts also apply when opioids are used in combination with propofol for TIVA. (26)

- Fig.6 Volatile anesthetic minimum alveolar concentration (MAC) reduction by opioids: the prototype example of isoflurane and fentanyl. The solid curve is MAC; the dotted curves are the 95% confidence intervals (CIs) (see text for details). (Adapted from McEwan AI, Smith C, Dyar O, et al. Isoflurane minimum alveolar concentration reduction by fentanyl. Anesthesiology. 1993;78:864-869, used with permission.)
Special Populations
Hepatic Failure
Even though the liver is the metabolic organ primarily responsible for the biotransformation of most opioids, liver failure is usually not severe enough to have a major impact on opioid pharmacokinetics. Of course, the anhepatic phase of orthotopic liver transplantation is a notable exception to this general rule (also see Chapter organ transplantation). With ongoing drug administration, concentrations of opioids that rely on hepatic metabolism increase when the patient has no liver. Even after partial liver resection, an increase in the ratio of morphine glucuronides to morphine occurs, indicating a decrease in the rate of morphine metabolism.(27) Because remifentanil’s metabolism is completely unrelated to hepatic clearance mechanisms, its disposition is not affected during liver transplantation.(28)
Pharmacodynamic considerations can be important for opioid therapy in patients with severe liver disease. Patients with ongoing hepatic encephalopathy are especially vulnerable to the sedative effects of opioids. As a consequence, this drug class must be used with caution in this patient population.
Kidney Failure
Renal failure has implications of major clinical importance with respect to morphine and meperidine (see the following discussion on individual drugs). For the fentanyl congeners, the clinical importance of kidney failure is much less marked. Remifentanil’s metabolism is not impacted by kidney disease. (29)
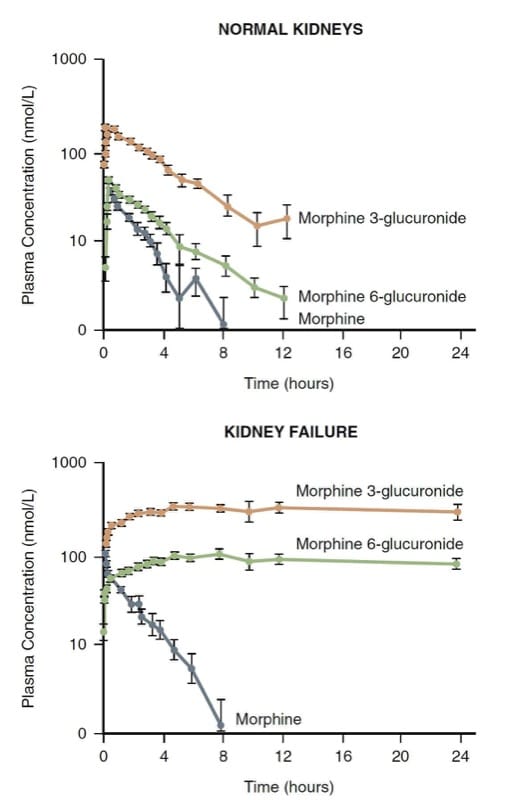
Morphine is principally metabolized by conjugation in the liver; the resulting water-soluble glucuronides (i.e., morphine 3-glucuronide and morphine 6-glucuronide—M3G and M6G) are excreted via the kidney. The kidney also plays a role in the conjugation of morphine and may account for as much as half of its conversion to M3G and M6G.
M3G is inactive, but M6G is an analgesic with a potency rivaling morphine. Very large levels of M6G and life-threatening respiratory depression can develop in patients with renal failure (Fig.7). (30) Consequently, morphine may not be a good choice in patients with severely altered renal clearance mechanisms.
The clinical pharmacology of meperidine is also significantly altered by renal failure. Normeperidine, the main metabolite, has analgesic and excitatory CNS effects that range from anxiety and tremulousness to myoclonus and frank seizures. Because the active metabolites are subject to renal excretion, CNS toxicity secondary to accumulation of normeperidine is especially a concern in patients with renal failure. This shortcoming of meperidine has caused many hospital formularies to restrict its use or to remove it from the formulary altogether.

- Fig.7 The pharmacokinetics of morphine and its metabolites in normal volunteers versus kidney failure patients. Note the significant accumulation of the metabolites in renal failure. (Adapted from Osborne R, Joel S, Grebenik K, et al. The pharmacokinetics of morphine and morphine glucuronides in kidney failure. Clin Pharmacol Ther. 1993;54:158-167, used with permission.)
Gender
Gender may have an important influence on opioid pharmacology. Morphine is more potent in women than in men and has a slower onset of action in women.(31) Some of these differences may be related to cyclic gonadal hormones and psychosocial factors.
Age
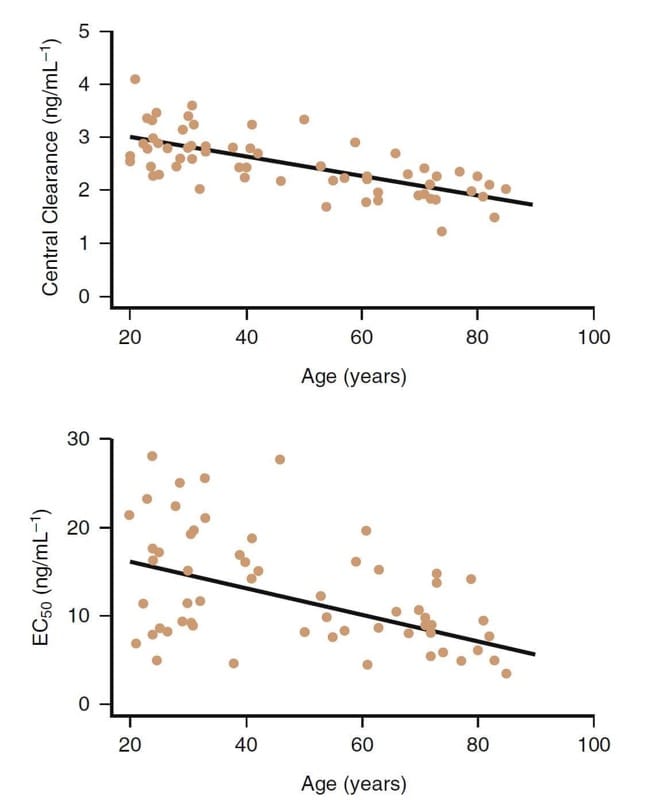
Advancing age is clearly an important factor influencing the clinical pharmacology of opioids. For example, fentanyl congeners are more potent in the older patient (Fig.8).(32,33) Decreases in clearance and central distribution volume also occur in older patients.
With advanced age, although pharmacokinetic changes also play a role, pharmacodynamic differences are primarily responsible for the decreased dose requirement in older patients (>65 years of age). Remifentanil doses should be decreased by at least 50% or more in elderly patients. Similar dosage reductions are also prudent for the other opioids as well.

- Fig.8 The influence of age on the clinical pharmacology of remifentanil. Although there is considerable variability, in general, older subjects have a lower central clearance and a higher potency (i.e., lower EC50).32
Obesity
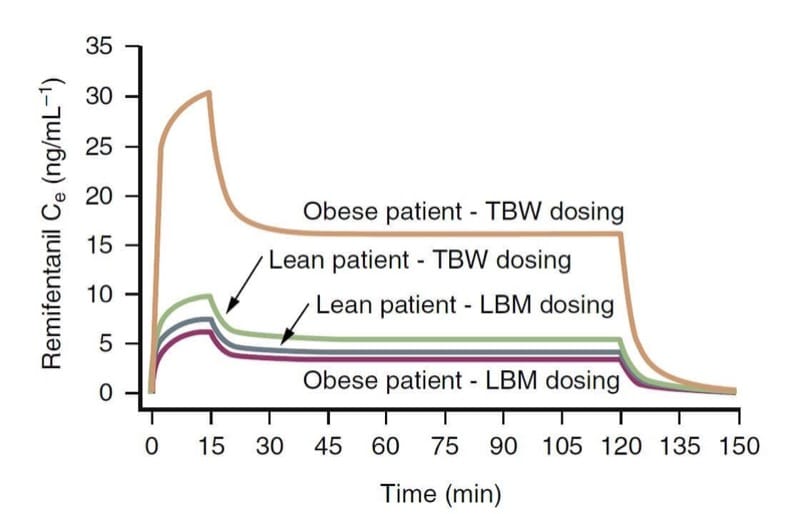
Body weight is likely an important factor influencing the clinical pharmacology of opioids. Opioid pharmacokinetic variables, especially clearance, are more closely related to lean body mass (LBM) rather than to total body weight (TBW). In practical terms, this means that morbidly obese patients do require a larger dosage than lean patients in order to achieve the same target concentration, but not as much as would be suggested by their TBW. (34)
For example, as illustrated through pharmacokinetic simulation (Fig. 9), a TBW-based dosing scheme results in much larger remifentanil effect-site concentrations than a dosing calculation based on LBM.(35) In contrast, TBW and LBM dosing schemes result in similar concentrations for lean patients. These concepts likely apply to other opioids as well.

- Fig.9 A pharmacokinetic simulation illustrating the consequences of calculating the remifentanil dosage based on total body weight (TBW) or lean body mass (LBM) in obese and lean patients (1 μg/kg bolus injection followed by an infusion of 0.5 μg/kg/min for 15 minutes and 0.25 μg/kg/min for an additional 105 minutes). Note that TBW-based dosing in an obese patient results in dramatically higher concentrations. (Adapted from Egan TD, Huizinga B, Gupta SK, et al. Remifentanil pharmacokinetics in obese versus lean patients. Anesthesiology. 1998;89:562-573, used with permission.)
Unique Features of Individual Opioids
Codeine
Codeine, although not commonly used intraoperatively, has special importance among opioids because of the well-characterized pharmacogenomic nuance associated with it. Codeine is actually a prodrug; morphine is the active compound. Codeine is metabolized (in part) by O-demethylation into morphine, a metabolic process mediated by the liver microsomal isoform CYP2D6.(36) Patients who lack CYP2D6 because of deletions, frame shift, or splice mutations (i.e., approximately 10% of the Caucasian population) or whose CYP2D6 is inhibited (e.g., patients taking quinidine) would not be expected to benefit from codeine even though they exhibit a normal response to morphine. (37,38)
Morphine
Morphine is the prototype opioid against which all newcomers are compared. There is no evidence that any synthetic opioid is more effective in controlling pain than nature’s morphine. Were it not for the histamine release and the resulting hypotension associated with morphine, fentanyl may not have replaced morphine as the most commonly used opioid intraoperatively.
Morphine has a slow onset time. Morphine’s pKa renders it almost completely ionized at physiologic pH. This property and its low lipid solubility account for morphine’s prolonged latency to peak effect; morphine penetrates the CNS slowly. This feature has both advantages and disadvantages associated with it. The prolonged latency to peak effect means that morphine is perhaps less likely to cause acute respiratory depression after bolus injection of typical analgesic doses compared to the more rapid-acting opioids. On the other hand, the slow onset time means that clinicians are perhaps more likely to inappropriately “stack” multiple morphine doses in a patient experiencing severe pain, thus creating the potential for a toxic “overshoot.”(39)
Morphine’s active metabolite, M6G, has important clinical implications. Although conversion to M6G accounts for only 10% of morphine’s metabolism, M6G may contribute to morphine’s analgesic effects even in patients with normal renal function, particularly with longer term use. Because of morphine’s high hepatic extraction ratio, the bioavailability of orally administered morphine is significantly lower than after parenteral injection. The hepatic first pass effect on orally administered morphine results in high M6G levels. In fact, M6G may be the primary active compound when morphine is administered orally.(40) As noted in the earlier section, “Kidney Failure,” M6G’s accumulation to potentially toxic levels in dialysis patients is another important implication of this active metabolite.
Fentanyl
Fentanyl may be the most important opioid used in modern anesthesia practice. As the original fentanyl congener, its clinical application is well entrenched and highly diverse. Fentanyl can be delivered in numerous ways. In addition to the intravenous route, fentanyl can be delivered by transdermal, transmucosal, transnasal, and transpulmonary routes.
Oral transmucosal delivery of fentanyl citrate (OTFC) results in the faster achievement of higher peak levels than when the same dose is swallowed. (41) Avoidance of the first pass effect results in substantially larger bio-availability. That OTFC is noninvasive and rapid in onset has made it a successful therapy for breakthrough pain in opioid-tolerant cancer patients, often in combination with a transdermal fentanyl patch (also see Chapter 40).
Alfentanil
Alfentanil was the first opioid to be administered almost exclusively by continuous infusion. Because of its relatively short terminal half-life, alfentanil was originally predicted to have a rapid offset of effect after termination of a continuous infusion.(42) Subsequent advances in pharmacokinetic knowledge (i.e., the CSHT) proved this assertion to be false.(8) However, alfentanil is in fact a short-acting drug after a single bolus injection because of its high “diffusible fraction”; it reaches peak effect-site concentrations quickly and then begins to decline (see the previous discussion of “Pharmacokinetics”). Alfentanil illustrates how a drug can exhibit different pharmacokinetic profiles depending upon the method of administration (i.e., bolus versus continuous infusion). Alfentanil, more than fentanyl or sufentanil, displays unpredictable hepatic metabolism because of the significant interindividual variability of hepatic CYP3A4, the primary enzyme responsible for alfentanil biotransformation.
Sufentanil
Sufentanil’s distinguishing feature is that it is the most potent opioid commonly used in anesthesia practice. Because it is more intrinsically efficacious at the opioid receptor, the absolute doses used are much smaller compared to the other less potent drugs (e.g., 1000-fold less than morphine doses).
Remifentanil
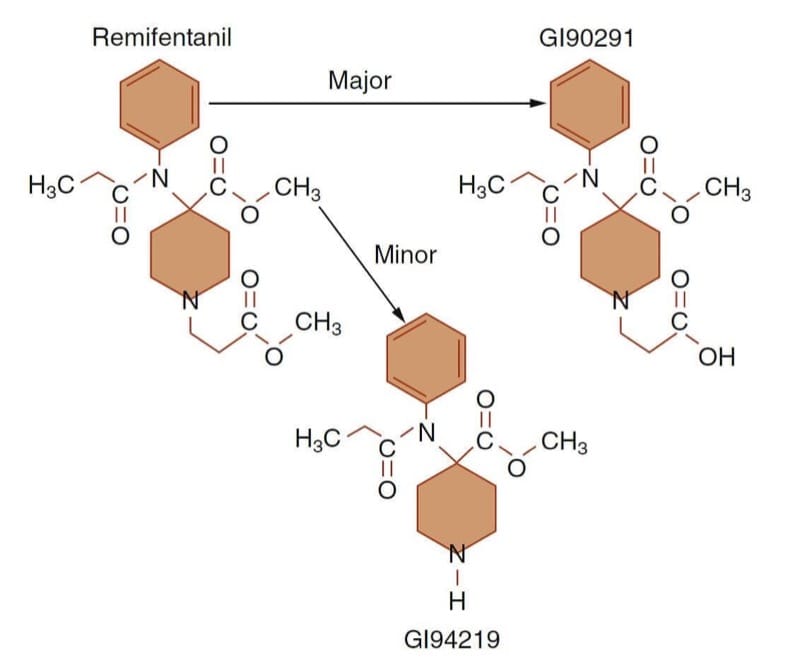
Remifentanil is a prototype example of how specific clinical goals can be achieved by designing molecules with specialized structure-activity (or structure-metabolism) relationships. By losing its μ-receptor agonist activity upon ester hydrolysis, a very short-acting opioid results (Fig.10). (43) The perceived unmet need driving remifentanil’s development was having an opioid with a rapid onset and offset so that the drug could be titrated up and down as necessary to meet the dynamic needs of the patient during the rapidly changing conditions of anesthesia and surgery.
Compared to the currently marketed fentanyl congeners, remifentanil’s CSHT is short, on the order of about 5 minutes. (44) Pharmacodynamically, remifentanil exhibits a short latency to peak effect similar to alfentanil and a potency slightly less than fentanyl. (45)
Remifentanil’s role in modern anesthesia practice is now relatively well established. Remifentanil is perhaps best suited for cases in which its responsive pharmacokinetic profile can be exploited to advantage (e.g., when rapid recovery is desirable; when the anesthetic requirement rapidly fluctuates; when opioid titration is unpredictable or difficult or when there is a substantial danger to opioid overdose; or when a “large dose” opioid technique is advantageous but the patient is not going to be mechanically ventilated postoperatively).(46) Remifentanil’s most common clinical application is the provision of TIVA in combination with propofol. It is also commonly administered by an intravenous bolus when only a very brief pulse of opioid effect followed by rapid recovery is desired (e.g., in preparation for local anesthetic injection during monitored anesthesia care) (see Chapter outpatient anesthesia).

- Fig.10 Remifentanil’s metabolic pathway. De-esterification (i.e., ester hydrolysis) by nonspecific plasma and tissue esterases to an inactive acid metabolite (GI90291) accounts for the vast majority of remifentanil’s metabolism. (Adapted from Egan TD, Huizinga B, Gupta SK, et al. Remifentanil pharmacokinetics in obese versus lean patients. Anesthesiology. 1998;89:562-573, used with permission.)
Opioid Agonist-Antagonists and Pure Antagonists

Opioid agonist-antagonists act as partial agonists at the μ-receptor, while having competitive antagonist properties at the same receptors. These drugs serve as analgesics with more limited ventilatory depression and a lesser potential for dependence as they demonstrate a “ceiling effect,” producing less analgesia compared to pure agonists. The lower abuse potential was the primary perceived unmet need underlying the development of these drugs. Drugs in this category are used for the treatment of chronic pain, as well as the treatment of opioid addiction. These drugs cause some degree of competitive antagonism when administered in the presence of ongoing full agonist activity (e.g., when administered after morphine and other pure agonists). Pure opioid antagonists, of which naloxone is the prototype, are complete competitive antagonists of the opioid receptor that are devoid of any agonist activity. These pure antagonists are used in the management of acute opioid overdose and chronic abuse.
Tramadol
Tramadol is a centrally acting analgesic with moderate μ-receptor affinity and weak κ- and δ-receptor affinity. Notably, tramadol also has antagonist activity at the 5-hydroxytryptamine (5-HT) and nicotinic acetylcholine (NA) receptors. While providing analgesia through both opioid and serotonin receptor pathways, tramadol carries less risk of respiratory depression. However, when combined with serotonin reuptake inhibitors or other serotonergic medications, it carries the risk of serotonin syndrome and also of CNS excitability and seizures.(47)
Buprenorphine
Buprenorphine is an opioid agonist-antagonist with a high affinity for the μ-receptor. It can be administered sublingually, transdermally, or parenterally but under-goes extensive first pass hepatic metabolism with oral administration. Although moderate doses can be used to treat chronic pain, higher doses used in the treatment of chronic pain can antagonize the effects of other opioids, making the treatment of acute on chronic pain difficult. Because it binds opioid receptors with such high affinity and its elimination half-life is in the range of 20 to 72 hours, large-dose opioid full agonists are required to overcome its effects.(48)
Nalbuphine
Also an opioid agonist-antagonist, nalbuphine has a potency and duration of action similar to morphine. It can be used as a sole drug for sedation with minimal respiratory depression, as well as a drug to reverse ventilatory depression in opioid overdose while maintaining some analgesia.(49)
Naloxone/Naltrexone
Naloxone is an injectable μ-antagonist that reverses both the therapeutic and adverse effects of μ-agonists.(50) Naloxone’s most common indication is the emergency reversal of opioid-induced ventilatory depression after acute overdose. Its important role in this regard has merited naloxone’s inclusion on the World Health Organization’s “List of Essential Medicines.” Naloxone is sometimes used in much smaller doses during emergence from anesthesia to restore adequate ventilatory effort and thereby expedite extubation of the trachea. The treatment of opioid-induced pruritus (requiring only small doses) is another common therapeutic application.
Although naloxone is very effective in reversing the ventilatory depression associated with opioids, it has numerous untoward effects, including acute withdrawal syndrome, nausea, vomiting, tachycardia, hypertension, seizures, and pulmonary edema, among others.(51) Recognizing that naloxone’s duration of action is shorter than that of most of the μ-agonists is a key point in determining the dosing schedule; repeated doses may be necessary to sustain its effects.
In response to the opioid abuse epidemic in the United States, new delivery systems have been developed that are intended for emergency use by laypersons in the event of opioid overdose; these include nasal spray and auto-injector preparations.(52,53)
Naltrexone, a longer acting opioid μ-antagonist available in oral, injectable, and implantable forms, is used in the long-term management of opioid addicts in combination with other nonpharmacologic therapies. (54)
3. CLINICAL APPLICATION
Opioids play a vital role in virtually every area of anesthesia practice. In the treatment of postoperative pain, opioids are of prime importance, whereas in most other settings in perioperative medicine opioids are therapeutic adjuncts used in combination with other drugs.
Common Clinical Indications
Postoperative analgesia is the longest standing indication for opioid therapy in anesthesia practice. In the modern era, opioid administration via PCA devices is perhaps the most common mode of delivery. In recent years, opioids are increasingly combined postoperatively with various other analgesics, such as nonsteroidal antiinflammatory drugs (NSAIDs), to increase efficacy and safety.
Internationally, the most common clinical indication for opioids in anesthesia practice is their use for what has come to be known as balanced anesthesia. This perhaps misguided term connotes the use of multiple drugs (e.g., volatile anesthetics, neuromuscular blockers, sedative-hypnotics, and opioids) in smaller doses to produce the state of anesthesia. With this technique, the opioids are primarily used for their ability to decrease MAC. A basic assumption underlying this balanced anesthesia approach is that the drugs used in combination mitigate the disadvantages of the individual drugs (i.e., the volatile anesthetics) used in larger doses as single drug therapy.
“Large-dose opioid anesthesia,” a technique originally described for morphine in the early days of open heart surgery (55) and later associated with the fentanyl congeners,(56) is another common application of opioids in clinical anesthesia. The original scientific underpinning of this approach was that large doses of opioids enabled the clinician to reduce the concentration of volatile anesthetic to a minimum, thereby avoiding the direct myocardial depression and other untoward hemodynamic effects in patients whose cardiovascular systems were already compromised. In addition, fentanyl often produces a relative bradycardia that could be helpful in patients with myocardial ischemia. Although the general concept is still applied, currently the opioid doses used are smaller. Opioids are also administered for their possible beneficial effects in terms of cardioprotection (i.e., preconditioning).
TIVA is a more recently developed and increasingly popular indication for opioids in anesthesia practice. This technique relies entirely upon intravenous drugs for the provision of general anesthesia. Most commonly, continuous infusions of remifentanil or alfentanil are combined with a propofol infusion. Both the opioid and the sedative are often delivered by target-controlled infusion (TCI) enabled pumps. A clear advantage of this technique, perhaps among others, is the enhanced patient well-being in the early postoperative period, including less nausea and vomiting and often a feeling of euphoria. (57)
Rational Drug Selection and Administration
In articulating a scientific foundation for rational opioid selection, pharmacokinetic considerations are extremely important. Indeed, the μ-agonists (opioids) can be considered pharmacodynamic equals with important pharmacokinetic differences.(58) Thus, rational selection of one opioid μ-agonist over another requires the clinician to identify the desired temporal profile of drug effect and then choose an opioid that best enables the clinician to achieve it (within obvious constraints such as pharmacoeconomic concerns).
In selecting the appropriate opioid, among the key questions to address are How quickly must the desired opioid effect be achieved? How long must the opioid effect be maintained? How critical is it that the opioid-induced ventilatory depression or sedation dissipate quickly (e.g., will the patient be mechanically ventilated postoperatively)? Is the capability to increase and decrease the level of opioid effect quickly during the anesthetic critical? Will there be significant pain postoperatively that will require opioid treatment? All of these questions relate to the optimal temporal profile of opioid effect. The answers to these questions are addressed through the application of pharmacokinetic concepts.
For example, when a brief pulse of opioid effect followed by rapid recovery is desired (e.g., to provide analgesia for a retrobulbar block), a bolus of remifentanil or alfentanil might be preferred. When long-lasting opioid effect is desired, such as when there will be significant postoperative pain or when the trachea will remain intubated, a fentanyl infusion is a prudent choice. If the patient should be awake and alert shortly after the procedure is finished (e.g., a craniotomy in which the surgeons hope to perform a neurologic examination in the operating room immediately postoperatively), a remifentanil infusion might be advantageous.
The formulation of a rational administration strategy also requires the proper application of pharmacokinetic principles. An important goal of any dosing scheme is to reach and maintain a steady-state level of opioid effect. Nowadays, in order to achieve a steady-state concentration in the site of action, opioids are frequently administered by continuous infusion. This is increasingly accomplished through the use of TCI technology, which requires that the clinician be familiar with the appropriate pharmacokinetic model for the opioid of interest. When these systems are not available, the clinician must remember that infusions must be preceded by a bolus in order to come to a near steady-state in a timely fashion.
4. EMERGING DEVELOPMENTS
Opioids and Cancer Recurrence
The influence of opioid therapy on cancer recurrence is controversial. As the immunosuppressive effects of opioids (particularly morphine) and their impact on angiogenesis have been demonstrated in animal and in vitro studies, concern over the influence of these drugs on cancer recurrence and survival has emerged. Some early retrospective data comparing cancer recurrence rates in patients receiving standard postoperative opioid analgesia with those receiving alternative techniques (e.g., epidural pain management) suggested a more frequent rate of cancer recurrence in the opioid therapy group; other studies found conflicting results. A retrospective review of more than 34,000 breast cancer patients from 1996 to 2008 demonstrated no association between opioid therapy and cancer recurrence. (59) Similarly, a retrospective review of 819 hepatocellular carcinoma patients who received either postoperative intravenous fentanyl or postoperative epidural with morphine found no effect on recurrence-free survival.(60)
However, other studies have suggested some improved outcomes with opioid-sparing techniques. A review of 984 non–small cell lung cancer patients from 2006 to 2011 found improved survival and longer disease-free survival in opioid-sparing pain management strategies. (61) Thus, the role of perioperative opioid therapy in cancer recurrence remains controversial; ongoing trials will further refine anesthesia-related clinical decision making in the treatment of oncologic patients.
Opioid Abuse Epidemic
Deaths related to the abuse and diversion of prescription opioids have skyrocketed in the United States and elsewhere. (62) In addition to fatalities, this pervasive pattern of prescription and illicit opioid abuse has resulted in a huge surge in admissions to opioid abuse treatment facilities. (63) The trend may be due at least in part to opioid prescribing practices for chronic pain conditions that may predispose some patients to addiction. (64,65)
The epidemic has reached such a crisis level that federal and state government authorities in the USA have enacted legislation and set aside funding to support research, prevention, and treatment of the problem. (66,67) State-approved pharmacy-based naloxone dispensing (without a physician’s prescription) for patients filling opioid prescriptions is a notable example of the efforts supported by such legislation. (68) In addition, professional societies and the Centers for Disease Control and Prevention (CDC) have produced new guidelines for opioid prescribing. (69) This is currently an area of intense public discussion and medical investigation.
5. QUESTIONS OF THE DAY
- A patient requires postoperative patient-controlled analgesia (PCA). From a pharmacokinetic perspective, what are the relative advantages of fentanyl compared to morphine for use in PCA?
- What pharmacokinetic parameter is most suitable for describing the offset time of a continuous opioid infusion?
- What are the effects of opioids on minute ventilation and ventilatory response to carbon dioxide?
- How does renal failure affect the pharmacokinetics of morphine and meperidine?
- A patient with postoperative respiratory depression from morphine is given intravenous naloxone. What are the potential side effects of naloxone?
- What key questions should be addressed when selecting an opioid for intraoperative use?