Nerve Blocks App
Nerve Blocks App Pain Medicine Assistant App
Pain Medicine Assistant App POCUS App
POCUS App IV Access App
IV Access App MSK Knee App
MSK Knee App VetRA App
VetRA App Nerve Block Manual
Nerve Block Manual Regional Anesthesia Updates
Regional Anesthesia Updates Anesthesiology Manual
Anesthesiology Manual Anesthesiology Review
Anesthesiology Review Anesthesia Updates 2025
Anesthesia Updates 2025 Anesthesia Updates 2026
Anesthesia Updates 2026 Pediatric Anesthesia Updates
Pediatric Anesthesia Updates Airway Management Updates
Airway Management Updates US Interventional Pain Manual
US Interventional Pain Manual Pain Medicine Updates
Pain Medicine Updates Mastering Difficult IV Access
Mastering Difficult IV Access PACU Nursing Manual
PACU Nursing Manual RA Veterinary Manual
RA Veterinary Manual About
About
Learning objectives
- Describe the causes and symptoms of myotonic dystrophy
- Diagnose and treat myotonic dystrophy
- Manage myotonic dystrophy patients presenting for surgery
Background
- Myotonic dystrophy (DM) is an autosomal dominant disorder characterized by muscle dystrophy starting in early adulthood
- Myotonic dystrophy type I (DM1, Steinert disease) and type II (DM2, proximal myotonic myopathy, milder form of type I)
- Multisystem disorder affecting somatic and smooth muscle, as well as ophthalmological, cardiovascular, endocrine, and central nervous systems
Etiology
- Genetic disorder caused by an expansion of DNA tandem repeats, resulting in an RNA gain of function mutation
- DM1 is caused by an expansion of a CTG repeat in the 3’-untranslated region of the DM1 protein kinase gene
- DM2 is caused by the expansion of a CCTG repeat in the intron of the CCHC-type zinc finger nucleic aced-binding protein gene
- DM is the most common muscular dystrophy in the European population
- DM1 is more common than DM2
Signs & symptoms
- Can range from potentially lethal in infancy to mild in late adulthood
- DM1 is classified into three types:
- Congenital myotonic dystrophy
- Fetal-onset involvement of muscle and central nervous system
- Reductions in fetal movement and polyhydramnios
- Equinovarus and ventriculomegaly on fetal ultrasound
- Neonatal mortality rate ~18%
- Childhood/adulthood: Characteristic tented appearance of the upper lip that results from facial diplegia, marked dysarthria, expressive aphasia, hypotonia rather dan myotonia
- Frequent respiratory involvement
- Mild myotonic dystrophy
- Mild muscle weakness, myotonia, and cataracts
- Onset between 20-70 years of age (typically after 40)
- Usually normal lifespan
- Classic myotonic dystrophy
- Onset during the second, third, or fourth decade of life
- Myotonia is the primary initial symptom
- Characterized by “warm-up phenomenon”: Symptoms appear more pronounced after rest and improve with muscle activity
- Distal muscle weakness is the main symptom, leading to impairment of fine motor tasks with the hands and impaired gait
- “Myopathic face”: due to weakness and wasting of facial, levator, palpebrae, and masticatory muscles
- Cardiac conduction abnormalities are common
- Reduced lifespan
- Congenital myotonic dystrophy
- DM2:
- Manifests in adulthood (median age 48 years) with a variable presentation
- Early-onset cataract, varying grip myotonia, proximal muscle weakness or stiffness, hearing loss, myofascial pain
- Weakness and/or myalgias are the most common initial symptoms
- Mostly axial and proximal muscle weakness affecting the neck flexors, long finger flexors, hip flexors, and hip extensors
- Abdominal, musculoskeletal, and exercise-related pain
- Sometimes misdiagnosed as fibromyalgia
Diagnosis
- Genetic testing
- Elevations in alkaline phosphatase, gamma-glutamyl transferase, serum aspartate aminotransferase, and serum alanine aminotransferase in 30-50% of patients
- Electrodiagnostic testing:
- Motor nerve conduction studies: Decreased amplitude with normal latency and normal conduction velocity
- Sensory nerve conduction studies: Typically normal
- Electromyography:
- Sustained runs of positive sharp waves
- Trains of negative spikes
- Fluctuating amplitude and frequencies
- Muscle biopsy: Type I fiber atrophy, Type 2 fiber hypertrophy, irregular fiber size, rows of internal nuclei, fibrosis, myofibrils oriented perpendicular to muscle fiber
Differential diagnosis
- Schwartz–Jampel Syndrome
- Duchenne muscular dystrophy
- Hyperkalemic Periodic Paralysis (HPP)
- Paramyotonia Congenita (PC)
- Myotonia Congenita
- Myotubular myopathy
- Acid maltase deficiency
- Debrancher deficiency
- Inflammatory myopathies
- Hypothyroid myopathy
- Chloroquine myopathy
- Statin myopathy
- Cyclosporine myopathy
Treatment
- No curative treatment, therapy is supportive and consists of monitoring and treating the issues associated with DM
| Cardiovascular | Annual ECG monitoring for cardiac conduction disturbances Baseline cardiac imaging every 1 to 5 years |
| Pulmonary | Obtaining baseline and serial pulmonary function testing to monitor for neuromuscular respiratory failure |
| Daytime somnolence and obstructive sleep apnea | Evaluate for sleep apnea and treat if necessary Consider neurostimulants (e.g., methylphenidate) for excessive sleepiness |
| Ocular involvement | Annual eye exam Surgical removal of cataracts |
| Obstetrics and gynecology | High-risk obstetrics evaluation for patients who are pregnant or considering pregnancy |
| Endocrine issues | Baseline and annual fasting blood glucose and hemoglobin A1C Screening for hypothyroidism Treat erectile dysfunction if necessary |
| Myotonia | Medications such as mexiletine, tricyclic antidepressants, benzodiazepines, or calcium antagonists reduce sustained myotonia Sodium channel blockers are contraindicated in patients with second and third-degree heart block |
| Muscle weakness | Physical and occupational therapy to strengthen muscles |
Complications
Central nervous system Intellectual disabilities
Cerebrovascular accidents
Anxiety and depression
Hypersomnia and sleep apnea
Ventriculomegaly
Ophtalmologic Cataracts
Hyperopia
Astigmatism
Cardiac Atrial arrhythmias
Conduction system slowing
Ventricular arrhythmias
Cardiomyopathy
Early-onset heart failure
Pulmonary Pneumonia
Increased risk of anesthesia-related pulmonary complications
Gastrointestinal Dysphagia
Gallstones and cholecystitis
Tranaminitis and liver enzyme elevations
Increased risk of post-anesthesia aspiration
Endocrine Insulin insensitivity
Testicular atrophy and male infertility
Increased risk of abortion, miscarriage, pre-term birth, dysmenorrhea
Dermatologic Androgenic alopecia
Increased risk of basal cell carcinoma and pilomatrixomas
Musculoskeletal Progressive loss of motor function
Myalgias
Anesthetic management
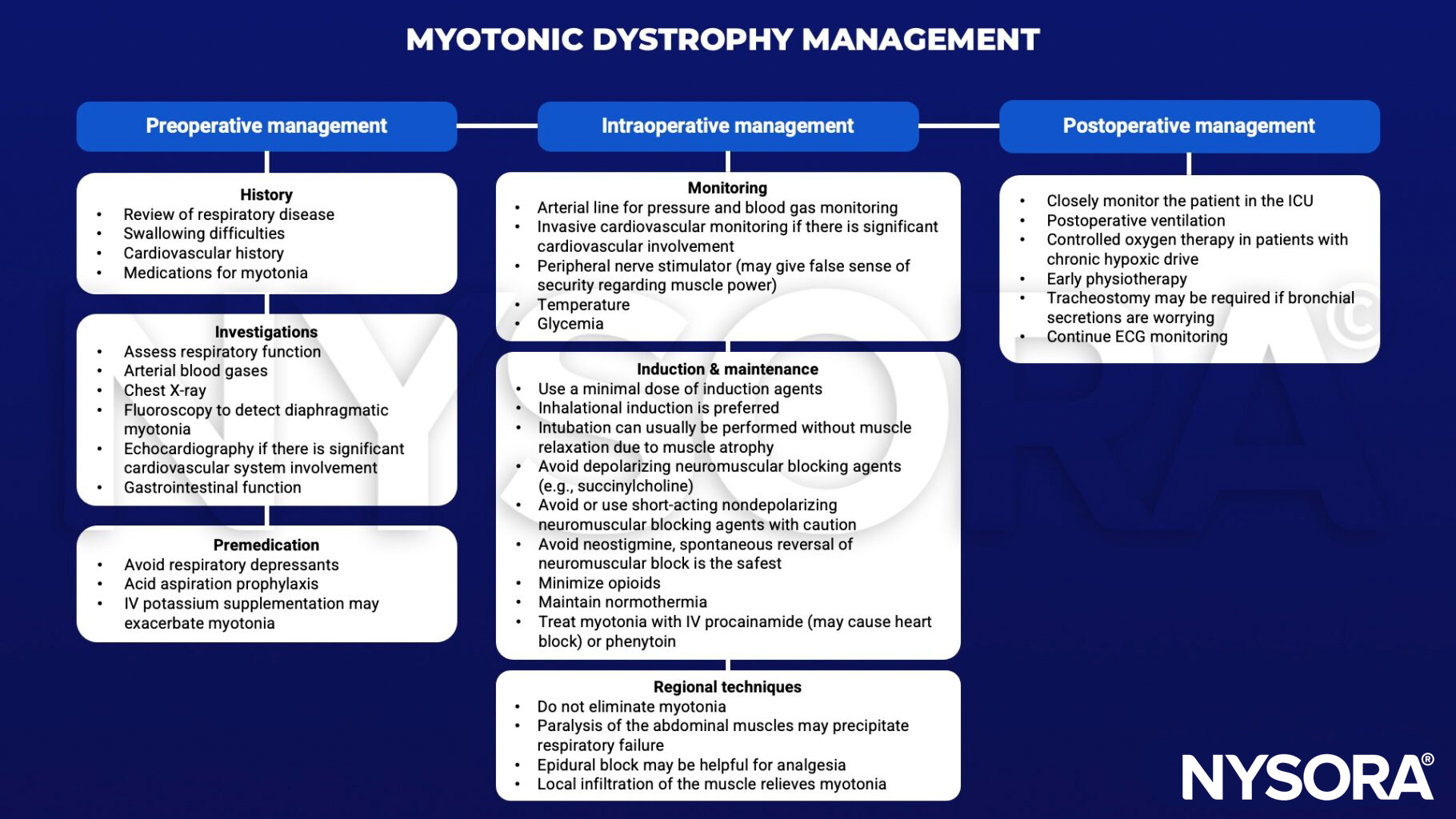
Suggested reading
- Vydra DG, Rayi A. Myotonic Dystrophy. [Updated 2022 Jun 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557446/
- Pollard BJ, Kitchen, G. Handbook of Clinical Anaesthesia. Fourth Edition. CRC Press. 2018. 978-1-4987-6289-2.
- Marsh S, Pittard A. Neuromuscular disorders and anaesthesia. Part 2: specific neuromuscular disorders. Continuing Education in Anaesthesia Critical Care & Pain. 2011;11(4):119-23.