INTRODUCTION
Local and regional anesthesia and analgesia appear to be undergoing a renaissance, as judged by attendance at specialty meetings and substantial increase in research activity, as evidenced by growing number of scientific publications. In contrast to general anesthesia, in which the molecular mechanism remains the subject of speculation, the site at which local anesthetic (LA) drugs bind to produce nerve blocks has been cloned and mutated. This chapter focuses on mechanisms of anesthesia and toxicity, especially as knowledge of these mechanisms will assist the clinician in conducting safer and more effective regional anesthesia.
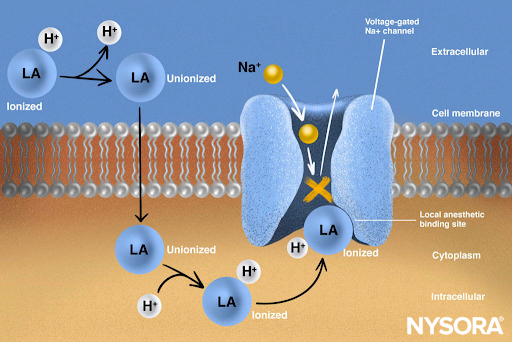
From the Regional Anesthesia Manual: Mechanism of action of local anesthetics. Local anesthetics work by binding to the α subunit of the voltage-gated Na+ channels, thus preventing the generation and conduction of nerve impulses. Subsequently, Na+ ions cannot flow into the cell, thereby halting the transmission of the advancing wave of depolarization down the length of the nerve. The fraction of local anesthetic molecules are in the ionized form. Local anesthetic molecules change from ionized to unionized in a fraction of a second.

From the Regional Anesthesia Manual: Mechanism of action of local anesthetics infographic.
PREHISTORY AND HISTORY
The Incas regarded coca as a gift from the son of the sun God and limited its use to the “upper crust” of society. They recognized and used the medicinal properties of cocaine long before the compound was brought to Europe for its properties to be “discovered.” The Incas sometimes treated persistent headaches with trepanation, and coca was occasionally used to facilitate this procedure. Local anesthesia was accomplished by having the operator chew coca leaves and apply the macerated pulp to the skin and wound edges while using a tumi knife to bore through the bone. By the sixteenth century, having disrupted Incan society, the conquistadors began paying laborers with cocaine paste.
The laborers generally rolled the cocaine leaves into balls (called cocadas), bound together by guano or cornstarch. These cocadas released the free-base cocaine as a consequence of the alkalinity of the guano and of the practice of chewing the cocadas with ash or lime (such alkaline compounds increase pH, favoring the free-base cocaine form over the positively charged hydrochloride salt). This practice probably marks the birth of “free-basing” cocaine and is the historic antecedent of the “rock” or “crack” cocaine so often abused in Western societies. Cocaine was brought back to Vienna by an explorer/physician named Scherzer. In Vienna, the chemist Albert Niemann isolated and crystallized pure cocaine hydrochloride in 1860. The Merck Company distributed batches of this agent to physicians for investigational purposes. Sigmund Freud was the most prominent of these cocaine experimenters. Freud reviewed his experimental work in a monograph devoted to cocaine, Über Coca. Freud and Carl Koller (an ophthalmology trainee) took cocaine orally and noticed that the drug rendered their tongues insensible. Koller and Joseph Gartner began a series of experiments using cocaine to produce topical anesthesia of the conjunctiva.
The birth of local and regional anesthesia dates from 1884, when Koller and Gartner reported their success at producing topical cocaine anesthesia of the eye in the frog, rabbit, dog, and human. The use of local anesthesia quickly spread around the world. The American surgeon William Halsted at Roosevelt Hospital in New York reported using cocaine to produce mandibular nerve block in 1884 and to produce brachial plexus block less than a year later. These blocks were accomplished by surgically exposing the nerves, then injecting them under direct vision. Leonard Corning injected cocaine near the spine of dogs, producing what was likely the first epidural in 1885. Spina anesthesia with cocaine was first accomplished in 1898 by August Bier. Cocaine spinal anesthesia was used to treat cancer pain in 1898. Caudal epidural anesthesia was introduced in 1902 by Sicard and Cathelin. Bier described intravenous regional anesthesia in 1909. In 1911, Hirschel reported the first three percutaneous brachial plexus anesthesias. Fidel Pages reported using epidural anesthesia for abdominal surgery in 1921. Cocaine was soon incorporated into many other products, including the original formulation of Coca-Cola devised by Pemberton in 1886. Wine tonics and other “patent” medicines of the day commonly contained cocaine (Figure 1). This practice ended when cocaine became regulated by the forerunner of the Food and Drug Administration (FDA) in the early 1900s.
")
FIGURE 1. Examples of products that incorporated cocaine during the time before it became a controlled substance. Wines fortified with cocaine were particularly popular as “tonics.” (Used with permission from Addiction Research Unit, University of Buffalo.)
MEDICINAL CHEMISTRY
Cocaine and all other LAs contain an aromatic ring and an amine at opposite ends of the molecule, separated by a hydrocarbon chain, and either an ester or an amide bond (Figure 2). Cocaine, the archetypical ester, is the only naturally occurring LA. Procaine, the first synthetic ester LA, was introduced in 1904 by Einhorn. The introduction of the amide LA lidocaine in 1948 was transformative. Lidocaine quickly became used for all forms of regional anesthesia. Other amide LAs based on the lidocaine structure (prilocaine, etidocaine) subsequently appeared. A related series of amide LAs based on 2’,6’-pipecoloxylidide was introduced (mepivacaine, bupivacaine, ropivacaine, and levobupivacaine). Ropivacaine and levobupivacaine are the only commercially available single-enantiomer (single-optical-isomer) LAs. Both are S(–)-enantiomers, avoiding the increased cardiac toxicity associated with racemic mixtures and the R(+)-isomers (this is discussed in a subsequent section). All other LAs either exist as racemates or have no asymmetric carbons.
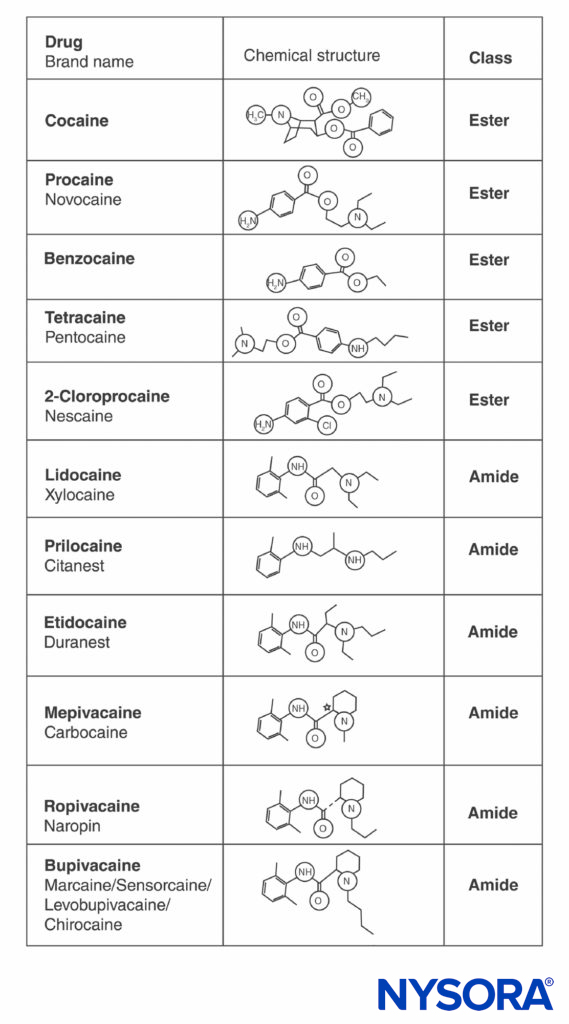
FIGURE 2. Structures of commonly used local anesthetics.
NYSORA Tips
- All LAs contain an aromatic ring and an amine at opposite ends of the molecule, separated by a hydrocarbon chain, and either an ester or an amide bond.
BIOPHYSICS OF VOLTAGE-GATED SODIUM CHANNELS AND LOCAL ANESTHETICS
Studies of the mechanisms of LA action on peripheral nerves are studies of interactions between LAs and voltage-gated Na channels because Na channels contain the LA-binding site. Na channels are integral membrane proteins that initiate and propagate action potentials in axons, dendrites, and muscle tissue; initiate and maintain membrane potential oscillations in specialized heart and brain cells; and shape and filter synaptic inputs. Na channels share structural features with other similar voltage-gated ion channels that exist as tetramers, each with six transmembrane helical segments (eg, voltage-gated Ca and K channels). Na channels contain one larger α-subunit and one or two smaller β-subunits, depending on the species and the tissue of origin. The α-subunit, the site of ion conduction and LA binding, has four homologous domains, each with six α-helical membrane-spanning segments (Figure 3). The external surface of the α-subunit is heavily glycosylated, which serves to orient the channel properly within the plasma membrane (Figure 4). Invertebrates have only one or two Na channel α-subunit genes, and the normal physiologic role of these channels is unclear (animals survive when the channels are not present).
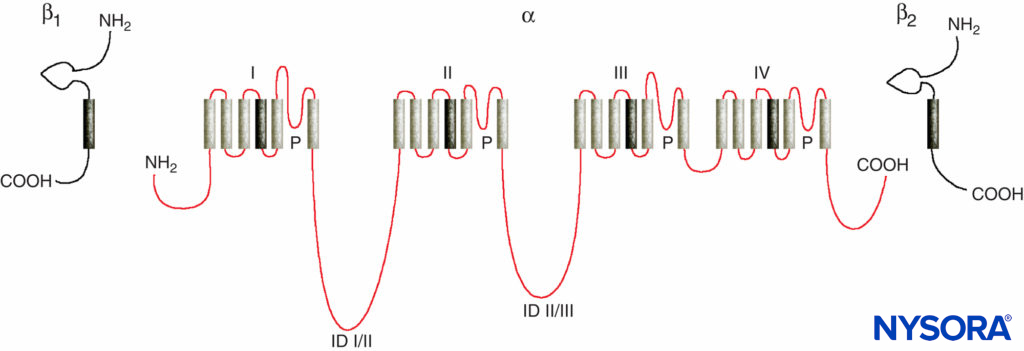
FIGURE 3. “Cartoon” structure of Na channel subunits. Note that the α-subunit has four domains that each contain six membrane-spanning segments.
. In contrast to local anesthetics, note that both scorpion toxins (ScTX) and tetrodotoxin (TTX) have binding sites on the external surface of the channel. Note also that the cytoplasmic side of the channel is phosphorylated. (Reproduced with permission from Catterall WA: Cellular and molecular biology of voltage-gated sodium channels. Physiol Rev. 1992 Oct;72(4 Suppl):S15-S48.)")
FIGURE 4. Cartoon of a Na channel in the plasma membrane. Note that all three subunits are heavily glycosylated on the extracellular side (see “squiggly” lines). In contrast to local anesthetics, note that both scorpion toxins (ScTX) and tetrodotoxin (TTX) have binding sites on the external surface of the channel. Note also that the cytoplasmic side of the channel is phosphorylated.
TABLE 1. Voltage-gated Na channel—neural isoforms.
| Nav 1.1 | Nav 1.2 | Nav 1.3 | Nav 1.6 | Nav 1.7 | Nav 1.8 | Nav 1.9 | |
|---|---|---|---|---|---|---|---|
| Chromosome | 2 | 2 | 2 | 12 | 2 | 3 | 3 |
| Where identified | CNS, DRG | CNS | CNS upregulated, after injury | DRG (large and small), CNS, Ranvier | DRG (large and small) | DRG (small) | DRG (small) |
| Inactivation | Fast | Fast | Fast | Fast | Fast | Slow | Slow |
| TTX | Sensitive | Sensitive | Sensitive | Sensitive | Sensitive | Insensitive | Insensitive |
Source: Adapted with permission from Novakovic SD, Eglen RM, Hunter JC: Regulation of Na+ channel distribution in the nervous system. Trends Neurosci. 2001 Aug;24(8):473-478.
Humans, in contrast, have nine active Na channel α-subunit genes on four chromosomes, with cell-specific expression and localization of gene products.10 The Nav 1.4 gene (by convention, geneticists refer to voltage-gated Na channel isoforms as Nav 1.x) supplies channels to skeletal muscle, and the Nav 1.5 gene supplies channels to cardiac muscle, leaving seven Nav isoforms in neural tissue (Table 1). Defined genes contribute specific Na channel forms to each of the unmyelinated axons, nodes of Ranvier in motor axons, and small dorsal root ganglion nociceptors. Whereas all Na channel α-subunits will bind LAs similarly, their affinity for binding neurotoxins varies. Na channel α- and β-subunit mutations lead to muscle, cardiac, and neural diseases. For example, inherited mutations in Nav 1.5 have been associated with congenital long QT syndrome, Bruguda syndrome, and other conduction system diseases. It has been shown that certain Nav isoforms proliferate in animal models of chronic pain. The existence of specific Nav gene α-subunit products offers the enticing possibility that inhibitors may someday be developed for each specific Nav α-subunit form. Such developments, already underway for some Nav isoforms, could revolutionize the treatment of chronic pain. Blocking of impulses in a nerve fiber requires that a defined length of nerve become inexcitable (to prevent the impulse from “jumping over” the blocked segment). Thus, as the LA concentration increases, it must be applied along a shorter length of nerve to prevent impulse conduction, as is shown in Figure 5. Both normal conduction and the way in which LAs inhibit conduction differ between myelinated and unmyelinated nerve fibers. Conduction in myelinated fibers proceeds in jumps from one Ranvier node to the next, a process termed saltatory conduction. To block impulses in myelinated nerve fibers, it is generally necessary for LAs to inhibit channels in three successive Ranvier nodes (Figure 6). Unmyelinated fibers, lacking the saltatory mechanism, conduct much more slowly than myelinated fibers. Unmyelinated fibers are relatively resistant to LAs, despite their smaller diameter, due to dispersal of Na channels throughout their plasma membranes. These differences among nerve fibers arise during development when Na channels begin to cluster at Ranvier nodes in myelinated axons. Nodal clustering of channels, essential for high-speed signal transmission, is initiated by Schwann cells in the peripheral nervous system and by oligodendrocytes in the central nervous system (CNS). Na channels may exist in at least three native conformations: “resting,” “open,” and “inactivated,” first described by Hodgkin and Huxley. During an action potential, neuronal Na channels open briefly, allowing extracellular Na ions to flow into the cell, depolarizing the plasma membrane. After only a few milliseconds, Na channels inactivate (where upon the Na current ceases). Na channels return to the resting conformation with membrane repolarization. The process by which channels go from conducting to nonconducting forms is termed gating. Gating is assumed to result from movements of dipoles in response to changes in potential. The process by which voltage-gated channels operate likely involves movements of paddle-shaped voltage sensors within the channel’s outer perimeter (Figure 7). The speed of gating processes differs among Nav α-subunit forms: Skeletal muscle and nerve forms gate quicker than cardiac forms.
:563-570.)")
FIGURE 5. Note that the concentration of local anesthetic required to produce nerve block declines as the length of nerve exposed to the local anesthetic increases.
:39-51.)")
FIGURE 6. Electron micrograph of a node of Ranvier. Na channels have been immunolabeled and appear as dense granules within the four arrows. The paranodal region is indicated by “pn,” and an astrocyte is indicated by “as.”
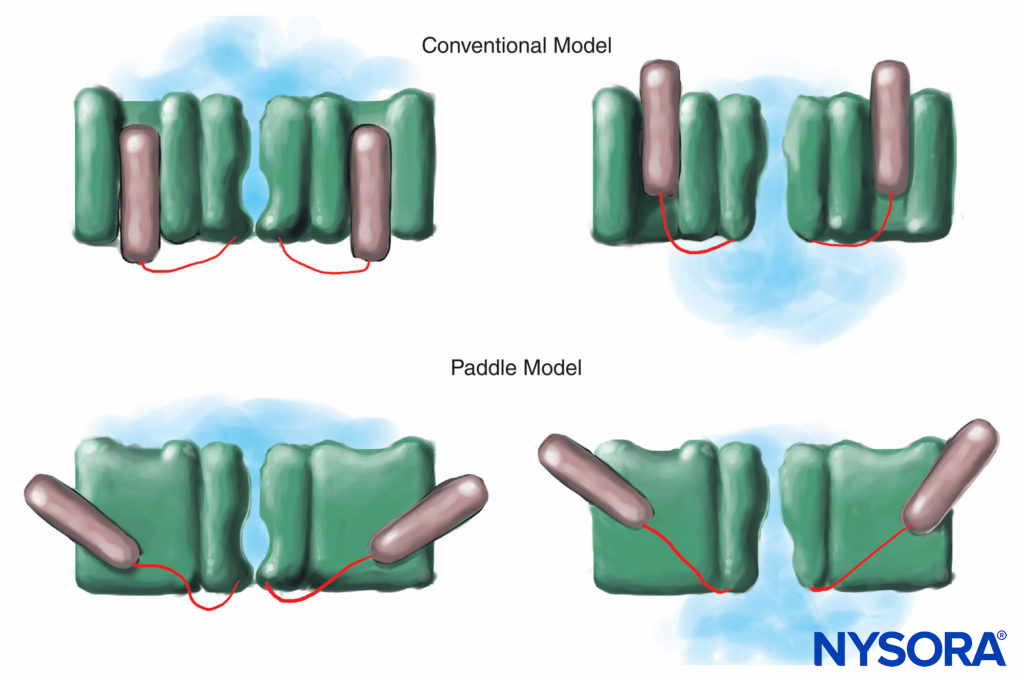
FIGURE 7. In the conventional model for voltage gating, the voltage-sensing part of the channel slides “in and out” of the membrane. More recent x-ray diffraction studies of the K channel suggest that a more appropriate mechanism is that of paddle-like structures sliding diagonally through the plasma membrane.
Anesthesia results when LAs bind Na channels and inhibit the Na permeability that underlies action potentials. Our understanding of LA mechanisms has been refined by several key observations. Taylor confirmed that LAs selectively inhibit Na channels in nerves. Strichartz first observed use-dependent block with LAs, showing the importance of channel opening for LA binding. Use (or frequency) dependence describes how LA inhibition of Na currents increases with repetitive depolarizations (“use”). Repetitive trains of depolarizations increase the likelihood that an LA will encounter an Na channel that is open or inactivated, with both forms having greater LA affinity than do resting channels (Figure 8). Thus, membrane potential influences both Na channel conformation and Na channel affinity for LAs. Use-dependent block appears important for the functioning of LAs as antiarrhythmics and may also underlie the effectiveness of reduced LA concentrations in managing pain. Finally, using site-directed mutagenesis, Ragsdale and Wang localized LA binding to specific amino acids in D4S6 of Nav 1.2 and Nav 1.4. Some LA optical isomers confer greater apparent safety than their opposite enantiomer. For example, under voltage clamp, the R(+)-bupivacaine isomer more potently inhibits cardiac Na currents than the S(–)-bupivacaine (levobupivacaine) isomer (Figure 9). Many other types of chemicals will also bind and inhibit Na channels, including general anesthetics, substance P inhibitors, α2-adrenergic agonists, tricyclic antidepressants, and nerve toxins. Nerve toxins are currently undergoing animal and human testing as possible replacements for LAs.
, reflecting an accumulating block of Na channels, until a nadir is reached. (Reproduced with permission from Hanck DA, Makielski JC, Sheets MF: Kinetic effects of quaternary lidocaine block of cardiac sodium channels: a gating current study. J Gen Physiol. 1994 Jan;103(1):19-43.)")
FIGURE 8. Use-dependent block of Na currents in Purkinje fibers. Under control conditions, each of a train of impulses results in identical current tracings. In the presence of the local anesthetic QX222, the first impulse is nearly the same size amplitude as under control conditions. Each succeeding impulse is smaller (reduced peak INa), reflecting an accumulating block of Na channels, until a nadir is reached.
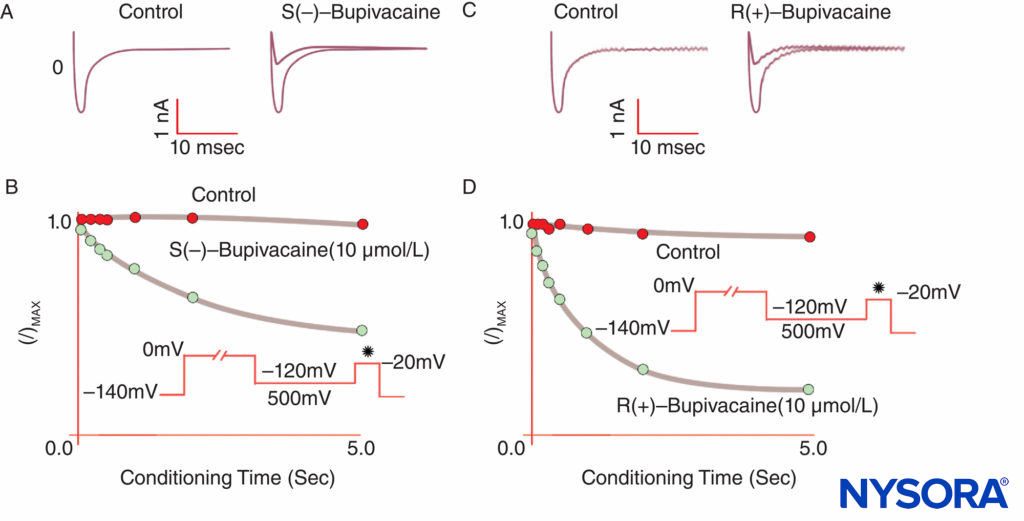
FIGURE 9. Reduced potency of S(–) bupivacaine relative to R(+)-bupivacaine at inhibiting cardiac Na currents under voltage clamp. After a standard “conditioning” depolarization of varying lengths, the S(–) isomer produces less reduction of I/Imax than the R(+) isomer.
LOCAL ANESTHETIC PHARMACODYNAMICS
In clinical practice, LAs are typically described by their potency, duration of action, speed of onset, and tendency for differential sensory nerve block. These properties do not sort independently.
Potency and Duration
Nerve-blocking potency of LAs increases with increasing molecular weight and increasing lipid solubility. Larger, more lipophilic LAs permeate nerve membranes more readily and bind Na channels with greater affinity. For example, etidocaine and bupivacaine have greater lipid solubility and potency than lidocaine and mepivacaine, to which they are closely related chemically.
NYSORA Tips
- Nerve-blocking potency of LAs increases with increasing molecular weight and increasing lipid solubility.
More lipid-soluble LAs are relatively water insoluble, highly protein bound in blood, less readily removed by the bloodstream from nerve membranes, and more slowly “washed out” from isolated nerves in vitro. Thus, increased lipid solubility is associated with increased protein binding in blood, increased potency, and longer duration of action. Extent and duration of anesthesia can be correlated with LA content of nerves in animal experiments. In animals, blocks of greater depth and longer duration arise from smaller volumes of more concentrated LA, compared with larger volumes of less-concentrated LA.
Speed of Onset
Many textbooks and review articles assert that the onset of anesthesia in isolated nerves slows with increasing LA lipid solubility and increasing pKa (Table 2). At any pH, the percentage of LA molecules present in the uncharged form, largely responsible for membrane permeability, decreases with increasing pKa.However, of the two LAs with the fastest onset, etidocaine is highly lipid soluble and chloroprocaine has a pKa greater than that of other LAs. Finally, the LA rate of onset is associated with the aqueous diffusion rate, which declines with increasing molecular weight.
TABLE 2. Local anesthetic characteristics that tend to sort together.
Physical and Chemical
- Increasing lipid solubility
- Increased protein binding
Pharmacologic and Toxicologic
- Increasing potency
- Increasing onset time
- Increasing duration of action
- Increasing tendency for severe systemic toxicity
- In general, all tend to sort together
Differential Sensory Nerve Block
Regional anesthesia and pain management would be transformed by a LA that would selectively inhibit pain transmission while leaving other functions intact. However, sensory anesthesia sufficient for skin incision usually cannot be obtained without motor impairment. As was first demonstrated by Gasser and Erlanger in 1929, all LAs will block smaller (diameter) fibers at lower concentrations than are required to block larger fibers of the same type. As a group, unmyelinated fibers are resistant to LAs compared with larger myelinated A-δ fibers. Bupivacaine and ropivacaine are relatively selective for sensory fibers. Bupivacaine produces more rapid onset of sensory than motor block, whereas the closely related chemical mepivacaine demonstrates no differential onset during median nerve blocks (Figure 10). True differential anesthesia may be possible when Nav isoform-selective antagonists become available. Certain Nav isoforms have been found to be prevalent in dorsal root ganglia, and (as previously noted) the relative populations of various Nav isoforms can change in response to various pain states.
, but not with mepivacaine 1% (mep). Note that the compound motor action potential (CMAP) is inhibited less than the sensory nerve action potential (SNAP) during onset of bupivacaine block in these normal volunteer subjects. At steady state (20 min), CMAP and SNAP are comparably inhibited. On the other hand, mepivacaine produced faster inhibition of both CMAP and SNAP, and there was no differential onset of block. (Reproduced with permission from Butterworth J, Ririe DG, Thompson RB, et al: Differential onset of median nerve block: randomized, double-blind comparison of mepivacaine and bupivacaine in healthy volunteers. Br J Anaesth. 1998 Oct;81(4):515-521.)")
FIGURE 10. Differential onset of median nerve block with bupivacaine 0.3% (bup), but not with mepivacaine 1% (mep). Note that the compound motor action potential (CMAP) is inhibited less than the sensory nerve action potential (SNAP) during onset of bupivacaine block in these normal volunteer subjects. At steady state (20 min), CMAP and SNAP are comparably inhibited. On the other hand, mepivacaine produced faster inhibition of both CMAP and SNAP, and there was no differential onset of block.
Other Factors Influencing Local Anesthetic Activity
Many factors influence the ability of a given LA to produce adequate regional anesthesia, including the dose, site of administration, additives, temperature, and pregnancy. As the LA dose increases, the likelihood of success and the duration of anesthesia increase, while the delay of onset and tendency for differential block decrease. In general, the fastest onset and shortest duration of anesthesia occur with spinal or subcutaneous injections; a slower onset and longer duration are obtained with plexus blocks.
NYSORA Tips
- The effectiveness of a given LA is influenced by the dose, site of administration, additives, temperature, and changes in neural susceptibility, as seen during pregnancy.
Epinephrine is frequently added to LA solutions to cause vasoconstriction and to serve as a marker for intravascular injection. Epinephrine and other α1-agonists increase LA duration largely by prolonging and increasing intraneural concentrations of LAs. Blood flow is decreased only briefly, and the block will persist long after the α1-adrenergic effect on blood flow has dissipated. Other popular LA additions include clonidine, NaHCO3, opioids, dexamethasone, and hyaluronidase. Uncharged local anesthetics have greater apparent potency at basic pH, where an increased fraction of LA molecules is uncharged, than at more acidic pH (Figure 11). Uncharged LA bases diffuse across nerve sheaths and membranes more readily than charged LAs, hastening onset of anesthesia. Some clinical studies showed that the addition of sodium bicarbonate had an inconsistent action during clinical nerve block; however, not all studies demonstrated a faster onset of anesthesia. One might anticipate that bicarbonate would have its greatest effect when added to LA solutions to which epinephrine was added by the manufacturer. Such solutions are more acidic than “plain” (epinephrine-free) LA solutions to increase shelf life. Bicarbonate shortens the duration of lidocaine in animals. Curiously, once LAs gain access to the cytoplasmic side of the Na channel, H+ ions potentiate use-dependent block. Marked prolongation of local anesthesia can be achieved by incorporating LAs into liposomes, as has been done with bupivacaine in the some formulation.
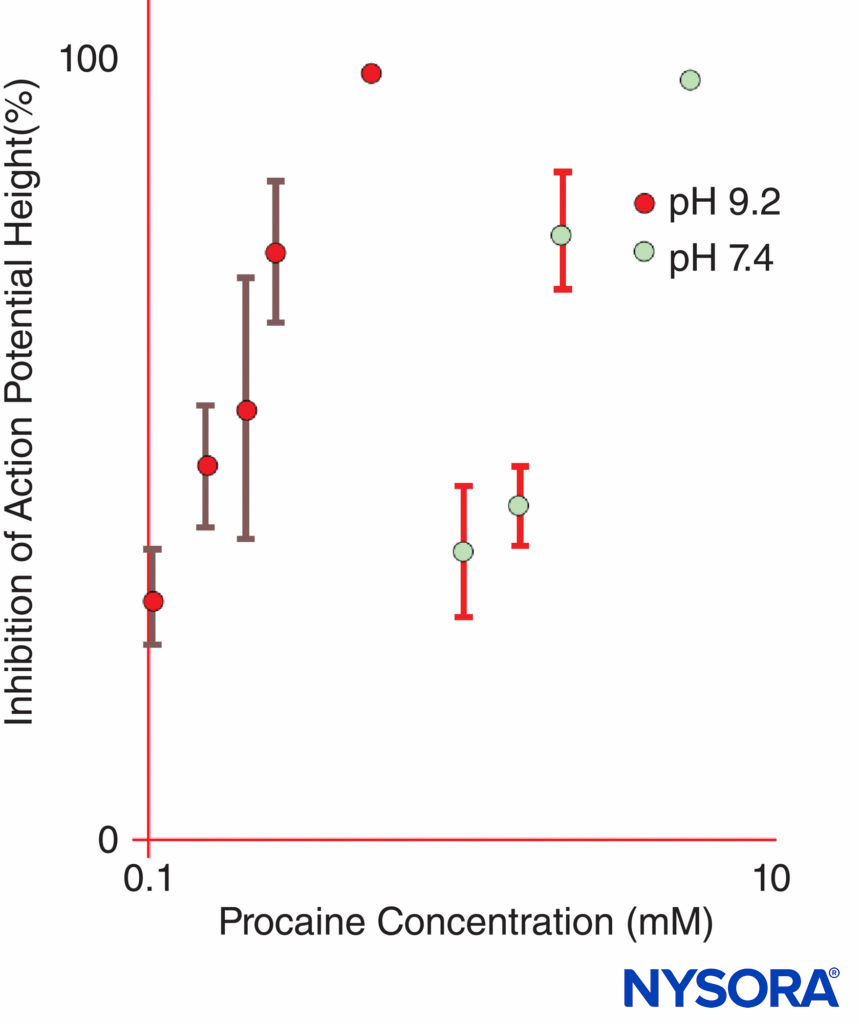
FIGURE 11. The potency of procaine at inhibiting compound action potentials in isolated frog sciatic nerves is dramatically increased at pH 9.2 as compared with pH 7.4.
NYSORA Tips
- Pregnancy increases neural susceptibility to LAs.
Pregnant women and pregnant animals demonstrate increased neural susceptibility to LAs. In addition, spread of neuraxial anesthesia likely increases during pregnancy due to decreases in thoracolumbar cerebrospinal fluid volume.
BLOOD CONCENTRATIONS AND PHARMACOKINETICS
Peak LA concentrations vary by the site of injection (Figure 12). With the same LA dose, intercostal blocks consistently produce greater peak LA concentrations than epidural or plexus blocks. As has been recently discussed by others, it makes little sense to speak of “maximal” doses of LAs except in reference to a specific nerve block procedure, since peak blood levels vary widely by block site. In blood, all LAs are partially protein bound, primarily to α1-acid glycoprotein and secondarily to albumin.
Affinity for α1-acid glycoprotein correlates with LA hydrophobicity and decreases with protonation (acidity). Extent of protein binding is influenced by the concentration of α1-acid glyco-protein. Both protein binding and protein concentration decline during pregnancy. During longer-term infusion of LA and LA-opioid combinations, concentrations of LA-binding proteins progressively increase There is considerable first-pass uptake of LAs by the lungs, and animal studies suggest that patients with right-to-left cardiac shunting may be expected to demonstrate LA toxicity after smaller intravenous bolus doses.
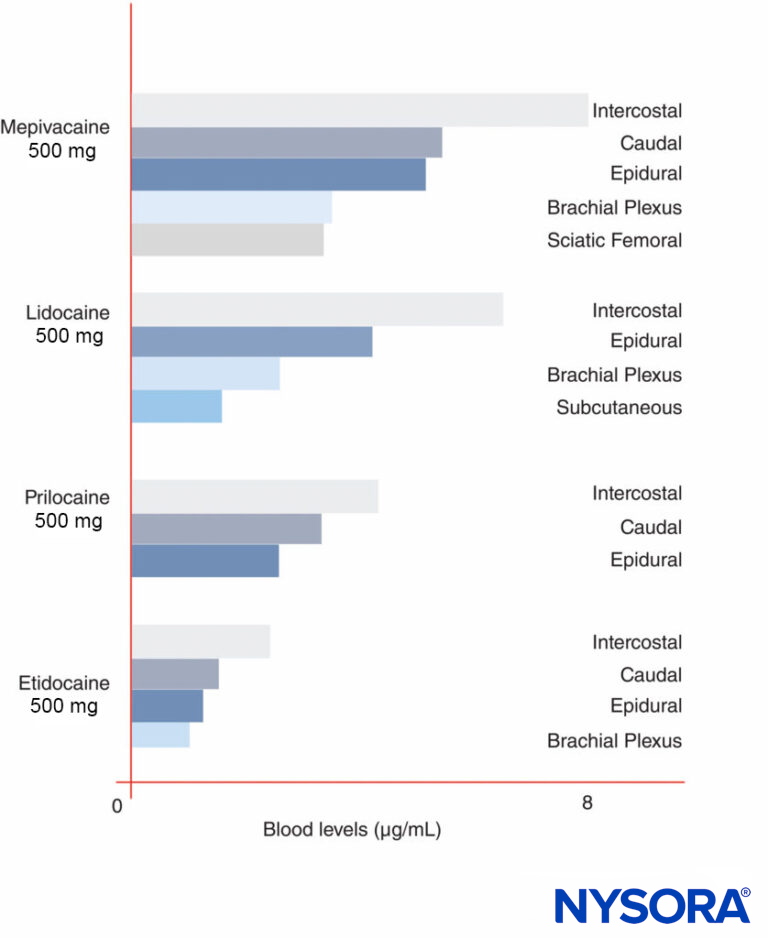
FIGURE 12. Peak blood concentrations of local anesthetics after various forms of regional anesthesia. Note that intercostal blocks consistently result in the greatest local anesthetic concentrations in blood, that plexus blocks result in the least local anesthetic concentrations in blood, and that epidural/caudal techniques are in between.
NYSORA Tips
- Recommendations on maximal doses of LAs commonly found in pharmacology texts are not terribly useful in the practice of clinical regional anesthesia.
- The serum concentrations of LAs depend on the injection technique, place of injection, and addition of additives to the LA.
- Any recommendation on the maximal safe LA dose can be valid only in reference to a specific nerve block procedure.
Esters undergo rapid hydrolysis in blood, catalyzed by non-specific esterases. Procaine and benzocaine are metabolized to para-aminobenzoic acid (PABA), the species underlying anaphylaxis to these agents. Higher doses of benzocaine, typically for topical anesthesia for endoscopy, can lead to life threatening levels of methemoglobinemia. The amides undergo metabolism in the liver. Lidocaine undergoes oxidative N-deal-kylation (by the cytochromes CYP 1A2 and CYP 3A4 to monoethyl glycine xylidide and glycine xylidide). Bupivacaine, ropivacaine, mepivacaine, and etidocaine also undergo N-dealkylation and hydroxylation. Prilocaine is hydrolyzed to o-toluidine, the agent that causes methemoglobinemia. Prilocaine doses of as little as 400 mg in fit adults may be expected to produce methemoglobinemia concentrations great enough to cause clinical cyanosis. Amide LA clearance is highly dependent on hepatic blood flow, hepatic extraction, and enzyme function; therefore, amide LA clearance is reduced by factors that decrease hepatic blood flow, such as β-adrenergic receptor or H2-receptor blockers, and by heart or liver failure. Disposition of amide LAs is altered in pregnancy due to increased cardiac output, hepatic blood flow, and clearance, as well as the previously mentioned decline in protein binding. Renal failure tends to increase volume of distribution of amide LAs and to increase the accumulation of metabolic by-products of ester and amide LAs. Theoretically, cholinesterase deficiency and cholinesterase inhibitors should increase the risk of systemic toxicity from ester LAs; however, there are no confirmatory clinical reports. Some drugs inhibit various cytochromes responsible for LA metabolism; however, the importance of cytochrome inhibitors varies depending on the specific LA species. β-Blockers and H2-receptor blockers inhibit CYP 2D6, which may contribute to reduced amide LA metabolism. Itraconazole has no effect on hepatic blood flow, but inhibits CYP 3A4 and bupivacaine elimination by 20%–25. Ropivacaine is hydroxylated by CYP 1A2 and metabolized to 2′,6′-pipecoloxylidide by CYP 3A4. Fluvoxamine inhibition of CYP 1A2 reduces ropiva-caine clearance by 70%. On the other hand, coadministration with strong inhibitors of CYP 3A4 (ketoconazole, itraconazole) has only a small effect on ropivacaine clearance.
DIRECT TOXIC SIDE EFFECTS
It is a common, but misguided, assumption that all LA actions, including toxic side effects, arise from interaction with voltage-gated Na channels. There is abundant evidence that LAs will bind many other targets aside from Na channels, including voltage-gated K and Ca channels, KATP channels, enzymes, N-methyl-D-aspartate receptors, β-adrenergic receptors, G-protein-mediated modulation of K and Ca channels, and nicotinic acetylcholine receptors. LA binding to any one or all of these other sites could underlie LA production of spinal or epidural analgesia and could contribute to toxic side effects.
Central Nervous System Side Effects
Local anesthetic CNS toxicity arises from selectively blocking the inhibition of excitatory pathways in the CNS, producing a stereotypical sequence of signs and symptoms as the LA concentration in blood gradually increases (Table 3). With increased LA doses, seizures may arise in the amygdala. With further LA dosing, CNS excitation progresses to CNS depression and eventual respiratory arrest. More potent (at nerve block) LAs produce seizures at lower blood concentrations and at lower doses than less-potent LAs. In animal studies, both metabolic and respiratory acidosis decreased the convulsive dose of lidocaine.
TABLE 3. Progression of signs and symptoms of toxicity as the local anesthetic dose (or concentration) gradually increases.
- Vertigo
- Tinnitus
- Ominous feelings
- Circumoral numbness
- Garrulousness
- Tremors
- Myoclonic jerks
- Convulsions
- Coma
- Cardiovascular collapse
Cardiovascular Toxicity
In laboratory experiments, most LAs will not produce cardiovascular (CV) toxicity until the blood concentration exceeds three times that necessary to produce seizures; however, there are clinical reports of simultaneous CNS and CV toxicity with bupivacaine (Table 4). In dogs, supraconvulsant doses of bupivacaine more commonly produce arrhythmias than supraconvulsant doses of ropivacaine and lidocaine. LAs produce CV signs of CNS excitation (increased heart rate, arterial blood pressure, and cardiac output) at lower concentrations than those associated with cardiac depression. Hypocapnia reduces ropivacaine-induced changes in ST segments and left ventricular contractility.
NYSORA Tips
- In laboratory experiments, most LAs will not produce CV toxicity until the blood concentration exceeds three times that necessary to produce seizures.
TABLE 4. Convulsive versus lethal doses of local anesthetics in dogs.
| Lidocaine | Bupivacaine | Tetracaine | |
|---|---|---|---|
| Dose producing convulsions in all animals (mg/kg) | 22 | 5 | 4 |
| Dose producing lethality in all animals (mg/kg) | 76 | 20 | 27 |
Local anesthetics bind and inhibit cardiac Na channels (Nav 1.5 isoform). Bupivacaine binds more avidly and longer than lidocaine to cardiac Na channels. As previously noted, certain R(+) optical isomers bind cardiac Na channels more avidly than S(–) optical isomers. These laboratory observations led to the clinical development of levobupivacaine and ropivacaine. Local anesthetics inhibit conduction in the heart with the same rank order of potency as for nerve block. Local anesthetics produce dose-dependent myocardial depression, possibly from interference with Ca signaling mech-anisms within cardiac muscle. These anesthetics bind and inhibit cardiac voltage-gated Ca and K channels at concentrations greater than those at which binding to Na channels is maximal. The LAs bind β-adrenergic receptors and inhibit epinephrine-stimulated cyclic adenosine monophosphate (AMP) formation. In rats, the rank order for cardiac toxicity appears to be bupivacaine > levobupivacaine > ropivacaine. In dogs, lidocaine was the least potent, and bupivacaine and levobupivacaine were more potent than ropivacaine at inhibiting left ventricular function as assessed by echocardiography (Table 5). In dogs, both programmed electrical stimulation and epinephrine resuscitation elicited more arrhythmias after bupivacaine and levobupivacaine than after lidocaine or ropivacaine administration. The mechanism by which CV toxicity is produced may depend on which LA has been administered. When LAs were given to the point of extreme hypotension, dogs receiving lidocaine could be resuscitated but required continuing infusion of epinephrine to counteract LA-induced myocardial depression. Conversely, many dogs receiving bupivacaine or levobupivacaine to the point of extreme hypotension could not be resuscitated. After bupivacaine, levobupivacaine, or ropivacaine, dogs that could be defibrillated often required no additional therapy. Similarly, in pigs, comparing lidocaine with bupivacaine, the ratio of potency for myocardial depression was 1:4, whereas it was 1:16 for arrhythmogenesis. The LAs produce dilation of vascular smooth muscle at clinical concentrations. Cocaine is the only LA that consistently produces local vasoconstriction.
Allergic Reactions
NYSORA Tips
- True immunologic reactions to LAs are rare.
- True anaphylaxis appears more common with ester LAs that are metabolized directly to PABA than other LAs.
- Accidental intravenous injections of LAs are sometimes misdiagnosed as allergic reactions.
- Some patients may react to preservatives, such as meth-ylparaben, included with LAs.
True immunologic reactions to LAs are rare. Accidental intravenous injections of LAs are sometimes misdiagnosed as allergic reactions. True anaphylaxis appears more common with ester LAs that are metabolized directly to PABA than to other LAs. Some patients may react to preservatives, such as methyl-paraben, included with LAs. Several studies have shown that patients referred for evaluation of apparent LA allergy, even after exhibiting signs or symptoms of anaphylaxis, almost never demonstrate true allergy to the LA that was administered. On the other hand, LA skin testing has an excellent negative predictive value. In other words, 97% of patients who fail to respond to LA skin testing will also not have an allergic reaction to the LA in a clinical setting.
TABLE 5. Efects of local anesthetics on indices of myocardial function measured in dogs.
| Local Anesthetic | LVEDP (EC50 for 125% base) (mcg/mL) | dP/dtmax (EC50 for 65% base) (mcg/mL) | %FS (EC50 for 65% base) (mcg/mL) |
|---|---|---|---|
| Bupivacaine | 2.2 (1.2–4.4) | 2.3 (1.7–3.1) | 2.1 (1.47–3.08) |
| Levobupivacaine | 1.7 (0.9–3.1) | 2.4 (1.9–3.1) | 1.3 (0.9–1.8) |
| Ropivacaine | 4.0 (2.1–7.5)a/sup> | 4.0 (3.1–5.2)b | 3.0 (2.1–4.2)a/sup> |
| Lidocaine | 6.8 (3.0–15.4)c | 8.0 (5.7–11.0)d | 5.5 (3.5–8.7)d |
Neurotoxic Effects
During the 1980s, 2-chloroprocaine (at that time formulated with sodium metabisulfite at a relatively acidic pH) occasion-ally produced cauda equina syndrome following accidental large-dose intrathecal injection during attempted epidural administration. Whether the “toxin” is 2-chloroprocaine or sodium metabisulfite remains unsettled: 2-chloroprocaine is now being tested as a substitute for lidocaine in human spinal anesthesia, and a series of publications suggest that it may be safe and effective. At the same time, other investigators have linked neurotoxic reactions in animals to large doses of 2-chlo-roprocaine rather than to metabisulfite. There is also contro-versy about transient neurologic symptoms and persistent sacral deficits after lidocaine spinal anesthesia. The reports and the controversy have persuaded many physicians to abandon lidocaine spinal anesthesia. Unlike other spinal LA solutions, lido-caine 5% permanently interrupts conduction when applied to isolated nerves or to isolated neurons. This may be the result of lidocaine-induced increases in intracellular calcium and does not appear to involve Na channel block. While it is impossible to “prove safety,” multiple studies suggest that chloroprocaine or mepivacaine can be substituted for lidocaine for brief spinal anesthesias.
Treatment of Local Anesthetic Toxicity
Treatment of adverse LA reactions depends on their severity. Minor reactions can be allowed to terminate spontaneously. Seizures induced by LAs should be managed by maintaining a patent airway and by providing oxygen. Seizures may be ter-minated with intravenous midazolam (0.05–0.10 mg/kg) or propofol (0.5–1.5 mg/kg) or a paralytic dose of succinylcho-line (0.5–1 mg/kg), followed by ventilation with bag and mask (or tracheal intubation). LA CV depression manifested by moderate hypotension, may be treated by infusion of intravenous fluids and vasopressors (phenylephrine 0.5–5 μg/kg/min, norepinephrine 0.02–0.2 μg/kg/min, or vasopressin 40 μg IV). If myocardial failure is present, epinephrine (1–5 μg/kg IV bolus) may be required. When toxicity progresses to cardiac arrest, the guidelines for treatment of LA toxicity as developed by the American Society of Regional Anesthesia and Pain Medicine (ASRA) are reasonable, and certainly preferable to the chaotic resuscitation schemes identified in a national survey prior to publication of the guideline. It makes sense that amiodarone be substituted for lidocaine and, based on multi-ple animal experiments, that smaller, incremental doses of epinephrine be used initially rather than 1-mg boluses. Animal experiments and clinical reports demonstrate the remarkable ability of lipid infusion to resuscitate from bupivacaine-induced cardiac arrest (Figure 13).Given the nearly nontoxic status of lipid infusion, one cannot make a convincing argument to withhold this therapy from a patient requiring resuscitation from LA intoxication. With unresponsive bupivacaine cardiac toxicity cardiopulmonary bypass should be considered. It appears that the threat from severe local anesthetic systemic toxicity may be on the decline, whether from better treatment or from changes in techniques. A minority would argue that the risk was overstated from the start, at least in experienced hands. Many practitioners believe that ultrasound guidance during peripheral nerve blocks has led to safer practices and less risk. While this view remains controversial, there are studies that support this belief.
 is given, but no arterial pressure is observed when CPR is discontinued. B: The same experiment is conducted, but a bolus of lipid is given; note that the arterial pressure is never lost (despite the same dose of bupivacaine being used), and that cardiac arrest does not ensue. (Reproduced with permission from Weinberg G: Current concepts in resuscitation of patients with local anesthetic cardiac toxicity. Reg Anesth Pain Med. 2002 Nov-Dec;27(6):568-575.)")
FIGURE 13. A: An anesthetized rat is given bupivacaine 15 mg/kg as indicated. The arterial blood pressure rapidly declines to cardiac arrest. Cardiopulmonary resuscitation (CPR) is given, but no arterial pressure is observed when CPR is discontinued. B: The same experiment is conducted, but a bolus of lipid is given; note that the arterial pressure is never lost (despite the same dose of bupivacaine being used), and that cardiac arrest does not ensue.
SUMMARY
After more than a century of use in Western medicine, LAs remain important tools for the twenty-first-century physician. Peripheral nerve blocks are almost certainly the result of LA inhibition of voltage-gated Na channels in neuronal membranes. The mechanisms of spinal and epidural anesthesia remain incompletely defined. The appropriate and safe dose of LAs varies with specific nerve block procedure. The mechanisms by which differing LAs produce CV toxicity likely vary: The more potent agents (eg, bupivacaine) may produce arrhythmias through a Na channel action, whereas the less-potent agents (eg, lidocaine) may produce myocardial depression through other pathways. Fears about LA systemic toxicity have abated with safer LAs, safer regional anesthesia practices, and improved treatments. There is renewed effort to produce clinically applicable delayed-release local anesthetic formulations to extend the duration of the currently available LAs.
Clinical updates
Taylor and McLeod (BJA Education, 2020) review the structure–function relationships of local anaesthetics, emphasizing that onset, potency, and duration are determined primarily by pKa, lipid solubility, and protein binding, respectively, and that differential fibre block (sympathetic > sensory > motor) reflects concentration-dependent sodium channel inhibition. They highlight clinically relevant pharmacokinetic distinctions—rapid plasma hydrolysis of esters versus hepatic metabolism of amides, additive systemic toxicity with cardiac NaV1.5 affinity highest for bupivacaine, and reduced cardiotoxicity with ropivacaine/levobupivacaine—while also summarizing emerging strategies such as liposomal formulations, toxin-derived agents (e.g., neosaxitoxin), and polymer-based extended-release systems aimed at prolonging analgesia without increasing toxicity.