Learning objectives
- Understanding of Glycogen storage disorders (GSD)
- Pre- and perioperative management of GSD
Definition and mechanisms
- Glycogen storage disorders (GSD) are metabolic disorders caused by an enzyme deficiency affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells
- Glycogen in skeletal muscles provides a ready source of fuel during exercise, and hepatic glycogen helps maintain plasma glucose levels during fasting
- Attempts to maintain glucose homeostasis can result in muscle degradation as amino acids are used as an alternative substrate
- Patients are at risk of hypoglycemia
- GSD is hereditary and occurs in about 1/20 000 babies
- There are 14 types of GS, with type I being the most common
Types of GSD
| Disease and eponym | Inheritance and incidence | Organs affected | Clinical features |
|---|---|---|---|
| Type I (von Gierke disease) | Autosomal recessive, 1:100 000–200 000 | Liver (and renal) | Hypoglycemia, lactic acidosis, ketosis, hepatomegaly, truncal obesity, short stature, hypertriglyceridemia, hyperuricemia and gout, platelet dysfunction, renal dysfunction Good prognosis with supportive treatment |
| Type II (Pompe disease) | Autosomal recessive, 1:50 000 | Cardiac, muscle, liver | Hypotonia and skeletal muscle weakness, hypertrophic cardiomyopathy. Death from cardiorespiratory failure by 2 yr in severe infantile form |
| Type III (Cori disease) | Autosomal recessive, 1:100 000–150 000 | Liver, muscles | hypoglycemia, ketonuria, hepatomegaly, muscle fatigue Good prognosis |
| Type IV (Andersen disease) | Autosomal recessive, 1:500 000 | Liver | Failure to thrive, hypotonia, hepatosplenomegaly Hepatic cirrhosis leading to death by 5 yr |
| Type V (McArdle disease) | Autosomal recessive, 1:500 000 | Muscles | Exercise intolerance, muscle cramps, fatigability Good prognosis |
| Type VI (Hers disease) | 1:200 000 | Liver | Hepatomegaly, mild hypoglycemia, hyperlipidemia, ketosis Good prognosis |
| Type VII (Tarui disease) | Autosomal recessive, 1:500 000 | Muscles | Similar to GSD type V |
| Type VIII/IX (phosphorylase kinase deficiency) | Autosomal and X-linked | Liver | Similar to GSD type III, but no myopathy |
Signs and symptoms
General symptoms of GSD may include:
- Poor growth
- Heat intolerance
- Bruising too easily
- Hypoglycemia
- An enlarged liver
- A swollen abdomen
- Low muscle tone
- Muscle pain and cramping during exercise
- Fatigue
- Obesity
- Kidney problems
- Low resistance to infections
- Mouth sores
- Heart problems
- Gout
Symptoms for babies may include:
- Too much acid in the blood (acidosis)
- High blood cholesterol levels (hyperlipidemia)
Diagnosis
- A low blood glucose level
- Abdominal ultrasound to detect an enlarged liver
- Tissue biopsy
- Abnormal blood tests
- Gene testing
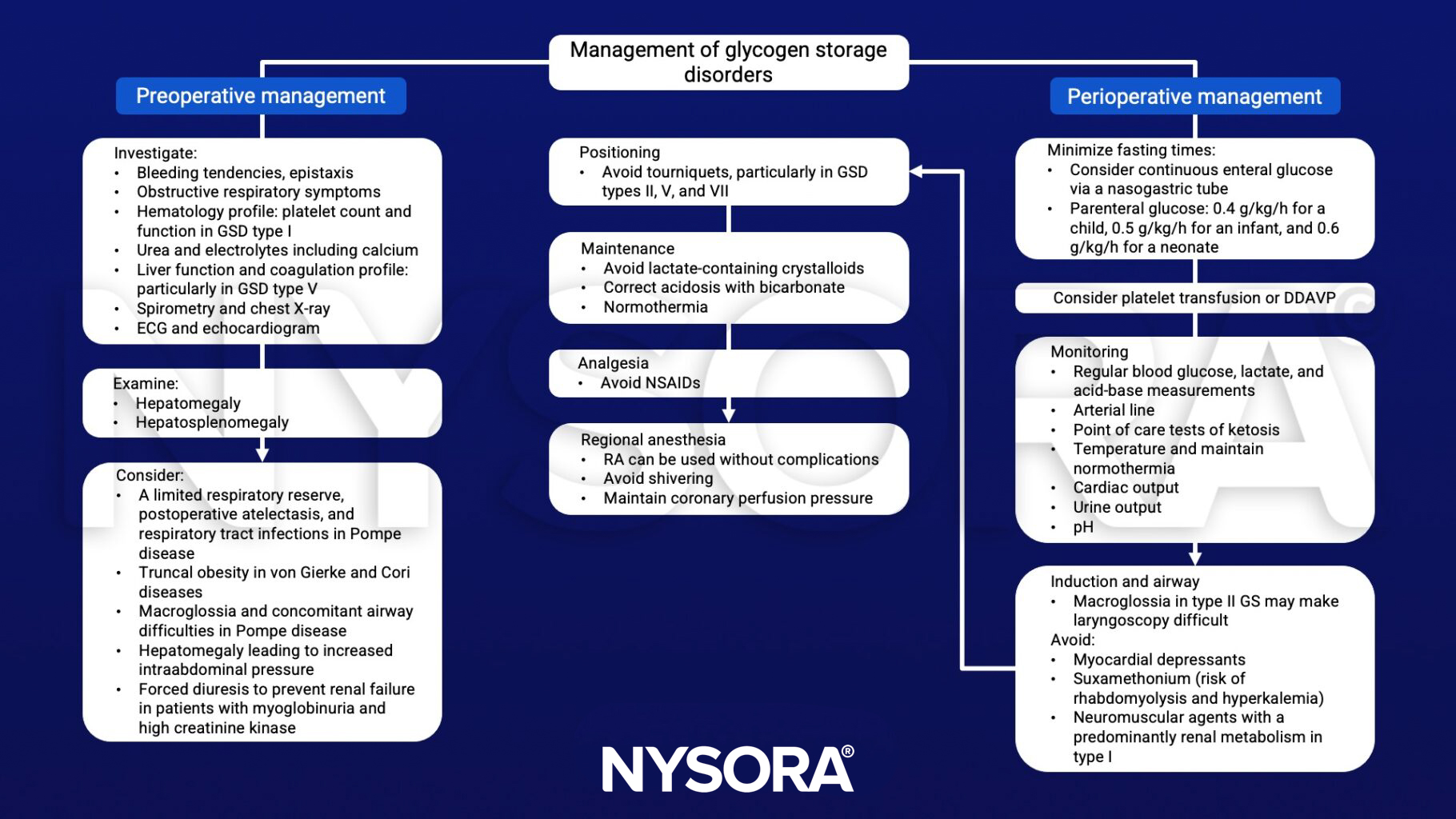
Management
Suggested reading
- Yeoh C, Teng H, Jackson J, et al. Metabolic Disorders and Anesthesia. Curr Anesthesiol Rep. 2019;9(3):340-359.
- Pollard BJ, Kitchen, G. Handbook of Clinical Anaesthesia. Fourth Edition. CRC Press. 2018. 978-1-4987-6289-2.
- Grant S, Nargis A. 2011. Perioperative care of children with inherited metabolic disorders. Continuing Education in Anaesthesia Critical Care & Pain.11:2;62-68.